




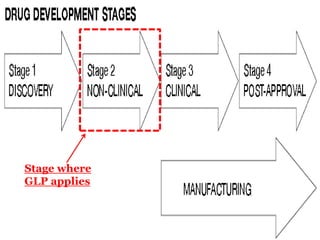
























































This document provides an overview of Good Laboratory Practice (GLP) standards. It defines GLP as a set of principles that provide a framework for conducting laboratory studies. GLP was established by the FDA in the 1970s after discovering fraudulent activities and poor practices in laboratories. GLP applies to nonclinical safety studies on chemicals, pharmaceuticals, and other products submitted to regulatory authorities. The document outlines the key components of GLP, including requirements for organization, personnel, facilities, equipment, testing operations, record keeping, and quality assurance. It also summarizes the various subparts and responsibilities defined in the 21 CFR part 58 GLP regulations.
















































































