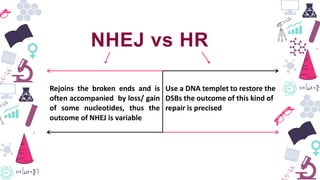
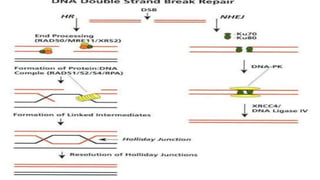
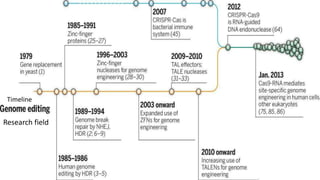
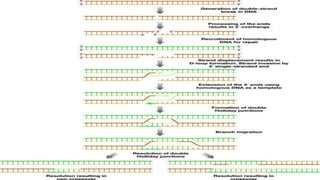
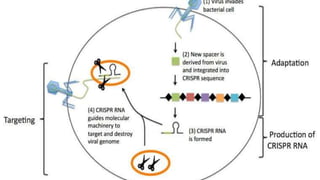
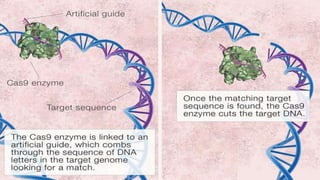
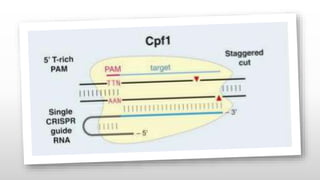
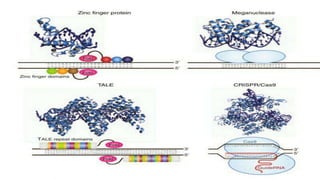
Genome editing is a technique used to precisely modify DNA within a cell. It involves using artificially engineered nucleases called "molecular scissors" to cut DNA at specific locations. This creates breaks that can then be repaired through natural cellular processes, allowing the genome to be altered. Early methods like homologous recombination were inefficient. New tools like zinc finger nucleases, TALENs, and the CRISPR/Cas9 system allow genome editing to be targeted to specific DNA sequences with greater accuracy and efficiency. These programmable nucleases make targeted cuts in the genome that can then be repaired through mechanisms like non-homologous end joining or homology-directed repair. CRISPR/Cas9 has become particularly






















































































![Polymer [ बहुलक ] Chemistry Notes PDF - Irfanullah Mehar - JJ Sir Chemistry.pdf](https://cdn.slidesharecdn.com/ss_thumbnails/polymerchemistrynotespdf-irfanullahmehar-jjsirchemistry-260210172118-3f9b37f7-thumbnail.jpg?width=640&height=640&fit=bounds)

















