Downloaded 1,760 times
![Next Gen Sequencing [NGS]
• History of DNA Sequencing
– Maxam-Gilbert
– Sanger
– ABI
• NGS Technologies:
– 454, Illumina, PacBio, ABI, Helicos,
– Ion Torrent, Nanopores
• Applications:
– Genomes, RNASeq, ChIPSeq, CGH, CancerGenome
, Environmental
Human Genome: 1990-2000
Presented by Dominic Suciu, Ph.D.](https://image.slidesharecdn.com/publicngs-talk-130710113342-phpapp02/85/Next-Gen-Sequencing-NGS-Technology-Overview-1-320.jpg)
![Next Gen Sequencing [NGS]
• History of DNA Sequencing
– Maxam-Gilbert
– Sanger
– ABI
• NGS Technologies:
– 454, Illumina, PacBio, ABI, Helicos,
– Ion Torrent, Nanopores
• Applications:
– Genomes, RNASeq, ChIPSeq, CGH, CancerGenome
, Environmental
Human Genome: 1990-2000
Presented by Dominic Suciu, Ph.D.](https://image.slidesharecdn.com/publicngs-talk-130710113342-phpapp02/75/Next-Gen-Sequencing-NGS-Technology-Overview-1-2048.jpg)
![Preliminaries: Central Dogma
Gene ~ Protein ~ Enzyme
Gene (DNA)
[Program in directory]
Protein (PolyPeptide)
[Program in RAM]
~~ Enzyme ~~
Functional agent
Messenger RNA
Genome (DNA)
[Hard drive]](https://image.slidesharecdn.com/publicngs-talk-130710113342-phpapp02/85/Next-Gen-Sequencing-NGS-Technology-Overview-2-320.jpg)
![Preliminaries: Phages
BacterioPhages are viruses that infect bacteria
Some Bacteria are immune to certain phages
[Hamilton O. Smith, early 70‟s]
Restriction Endonucleases: Enzymes that specifically
cleave certain DNA sequences.
Bacterial cells use these as a crude anti-phage
defense mechanisms](https://image.slidesharecdn.com/publicngs-talk-130710113342-phpapp02/85/Next-Gen-Sequencing-NGS-Technology-Overview-3-320.jpg)

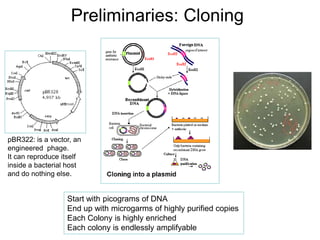
![Preliminaries: PCR [1985]
As long as you know
the beginning and
end of a sequence,
you can amplify
anything](https://image.slidesharecdn.com/publicngs-talk-130710113342-phpapp02/85/Next-Gen-Sequencing-NGS-Technology-Overview-6-320.jpg)



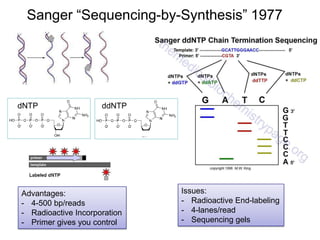
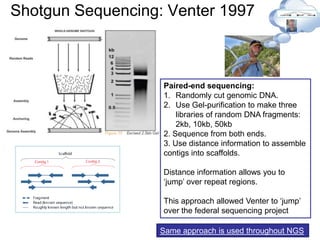
![Human Genome Project (15 years) Hierarchical
Shotgun Sequencing [start1990]
- Randomly insert Human DNA into BAC clones (~150kbp each)
- Combine these BAC clones to create a scaffold of the human
genome. Each BAC clone will be mapped to a region on a
Human Chromosome
- Pass BAC clones to different Genome Centers throughout US
- At each center, each vector is sequenced using shotgun sequencing
- Wait 15 years for results.](https://image.slidesharecdn.com/publicngs-talk-130710113342-phpapp02/85/Next-Gen-Sequencing-NGS-Technology-Overview-12-320.jpg)
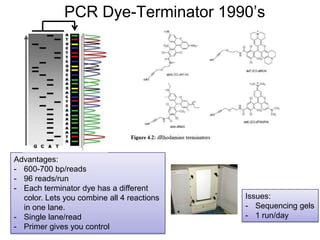
![NGS Revolution: Roche / 454 -> [2005]
ABI 3700 state of the art
in 1997
- 1 sample per rxn (96
rxns) in 2 hrs
- Each sample had to be
individually manipulated
454 solved both these problems
PPi + H+
Paired-end reads can be done by including both primers on each micro-bead
Emulsion PCR:](https://image.slidesharecdn.com/publicngs-talk-130710113342-phpapp02/85/Next-Gen-Sequencing-NGS-Technology-Overview-15-320.jpg)
![Roche / 454 -> [2005]
• emPCR: No need for
cells
• Each well is a single
sequencing run.
• Very fast reaction](https://image.slidesharecdn.com/publicngs-talk-130710113342-phpapp02/85/Next-Gen-Sequencing-NGS-Technology-Overview-16-320.jpg)
![Illumina [Solexa 2007]
No need for Cell-based
amplification
Bridge Amplification: PCR on
a surface](https://image.slidesharecdn.com/publicngs-talk-130710113342-phpapp02/85/Next-Gen-Sequencing-NGS-Technology-Overview-17-320.jpg)

![Ion Torrent/LifeTechnologies [2010]
Method:
• Emulsion PCR
• Each bead is placed in a
single well.
• CHEAP/Rugged
Disadvantages:
• Low density
• Sample prep
PPi + H+](https://image.slidesharecdn.com/publicngs-talk-130710113342-phpapp02/85/Next-Gen-Sequencing-NGS-Technology-Overview-19-320.jpg)
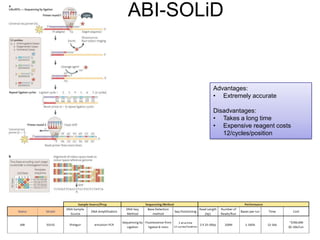
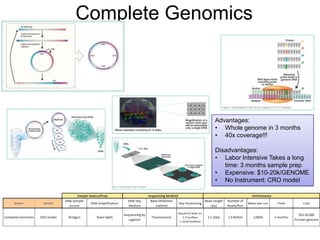
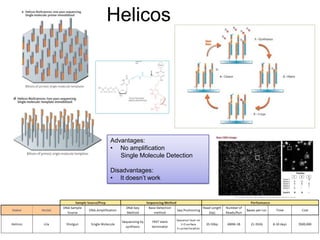
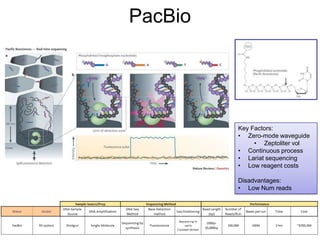
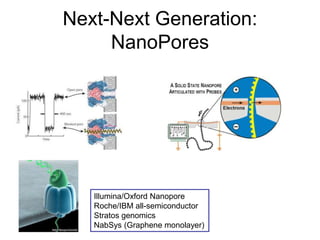





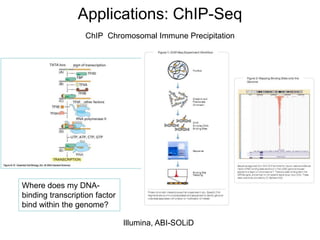
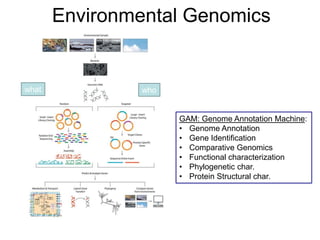
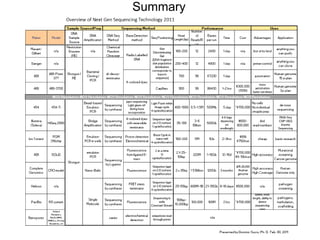

Next generation sequencing (NGS) provides several new technologies for DNA sequencing that have significantly increased throughput and reduced costs compared to previous methods. NGS technologies include Roche/454, Illumina, ABI SOLiD, Ion Torrent, and PacBio. These technologies have various applications including whole genome sequencing, detection of genetic mutations associated with diseases, RNA sequencing to study gene expression, and ChIP sequencing to identify DNA-binding sites. NGS is revolutionizing genomic research by allowing comprehensive study of genomes, transcriptomes, and gene regulation.


































































