Download as PDF, PPTX




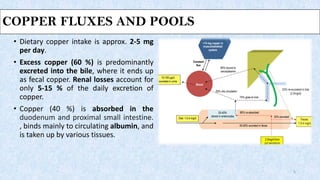
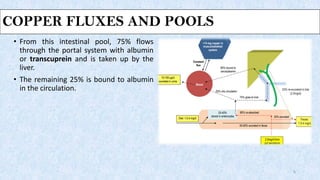






















This document discusses Wilson's disease, including its pathophysiology, clinical manifestations, diagnosis, and management. Some key points: 1. Wilson's disease is caused by a genetic defect impairing copper transport, leading to copper accumulation and toxicity in the liver and other organs. 2. Clinical manifestations can include liver disease (cirrhosis, acute liver failure), neurological symptoms, and psychiatric issues. Kayser-Fleischer rings seen in the cornea are also characteristic. 3. Diagnosis involves low serum ceruloplasmin levels (<20 mg/dL), elevated 24-hour urinary copper excretion (>40 mcg), and response to copper-chelating agents. 4






































![[Wilson disease][20170907][高醫附醫][intern陳佳菁]](https://cdn.slidesharecdn.com/ss_thumbnails/wilsondisease20170907intern-171115010736-thumbnail.jpg?width=640&height=640&fit=bounds)
















































