Downloaded 29 times






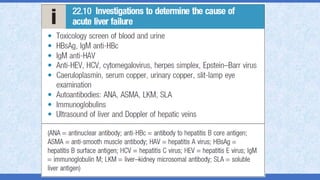



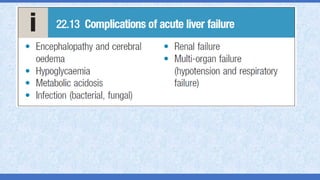

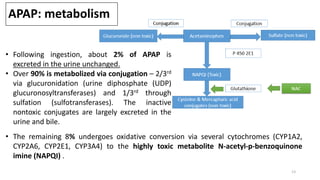
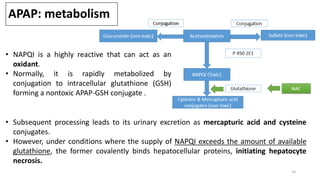









The document provides an overview of acute liver failure (ALF), including its definition, clinical assessment, causes, management strategies, and specific treatments for acetaminophen and mushroom poisoning. It outlines prognostic factors, the importance of intensive care unit treatment, and the critical role of N-acetylcysteine in acetaminophen overdose. Additionally, it discusses the phases of mushroom poisoning and treatment options for hepatic injury caused by Amanita species toxins.





![[2015] hepatic encephalopathy](https://cdn.slidesharecdn.com/ss_thumbnails/2015hepaticencephalopathy-151117004212-lva1-app6891-thumbnail.jpg?width=640&height=640&fit=bounds)

















































































