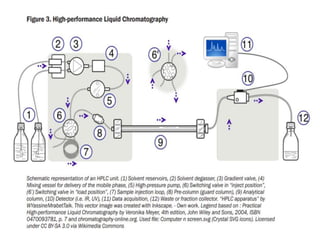
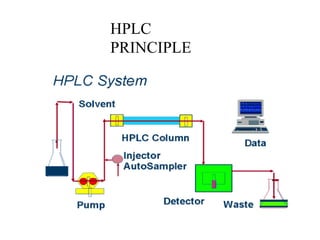
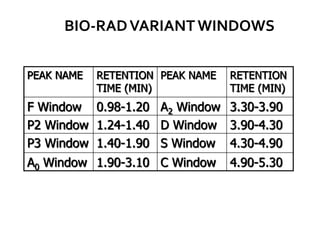
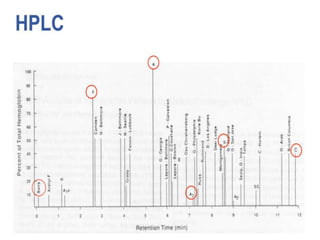
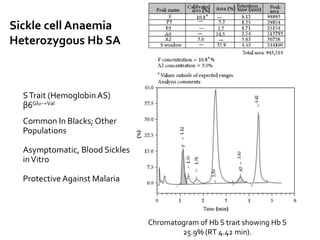
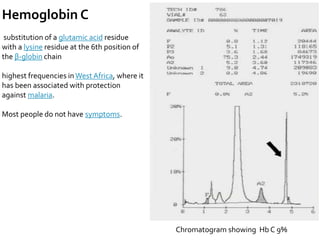
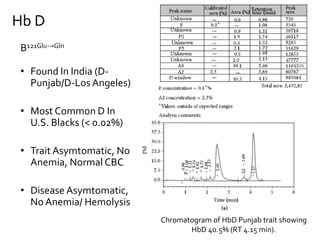
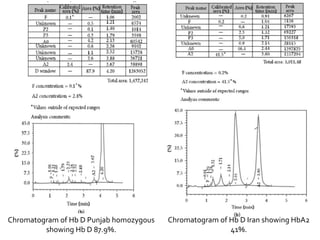
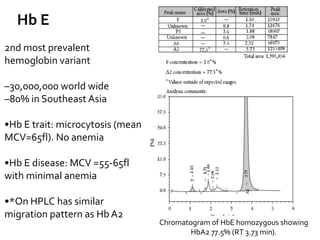
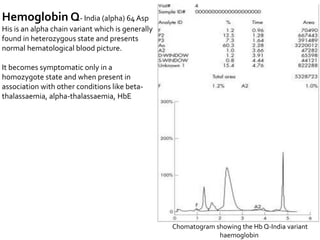
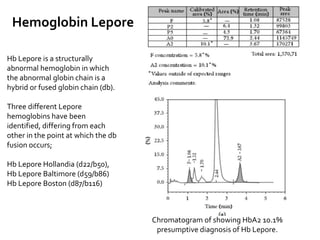
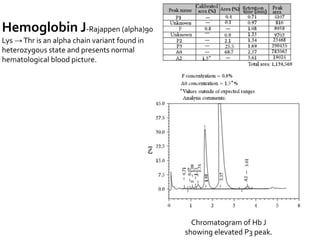
HPLC is used to separate and identify hemoglobin variants and diagnose hemoglobinopathies. It works by separating hemoglobin fractions based on their ionic interactions with a chromatographic cartridge using an increasing ionic strength buffer gradient. Each hemoglobin variant has a characteristic retention time. Abnormalities in the hemoglobin protein are called hemoglobinopathies, which can include sickle cell anemia, thalassemias, and over 800 known variants. The document provides examples of HPLC chromatograms and interpretations for various normal hemoglobin patterns and hemoglobinopathies.


















![Chromatogram of Hb Hope showing elevated
P2 peak (48.4%).
Hb Hopeis a clinically asymptomatic
β- chain variant [beta136 (H14) Gly→Asp
(GGT→GAT)].
It is more prevalent in Mediterranean region of
the world than in Asian countries and
extremely rare in India.
Causes Spuriously Elevated HbA1cValues on
HPLC Assay.](https://image.slidesharecdn.com/deepesh-240201132221-3799f99a/85/thalassemia-38-320.jpg)
















































![jodhpur presentation [Autosaved].pptx12 final copy1-1.pptx](https://cdn.slidesharecdn.com/ss_thumbnails/jodhpurpresentationautosaved-240131153020-1cfa8221-thumbnail.jpg?width=640&height=640&fit=bounds)





























