Download to read offline

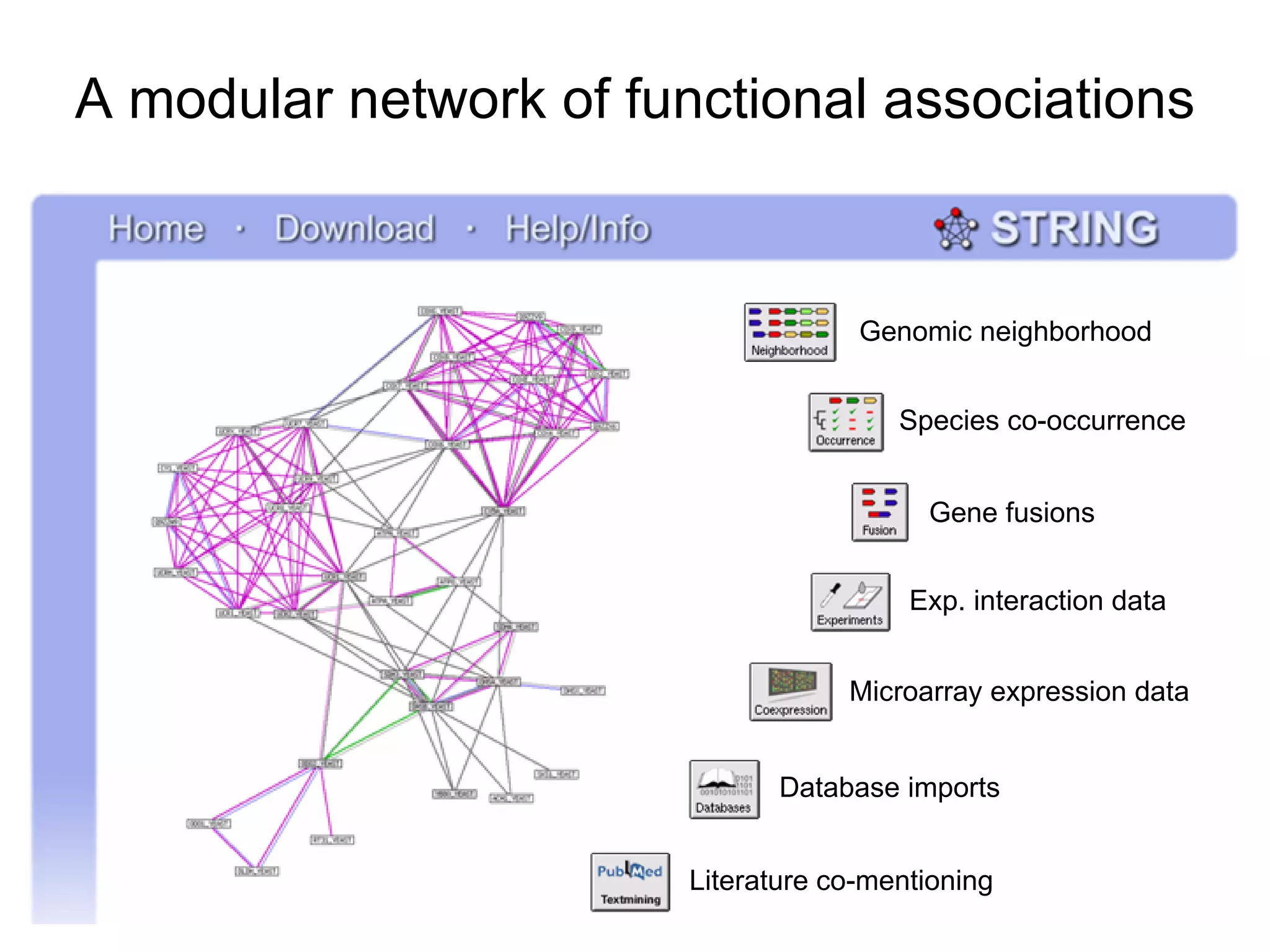
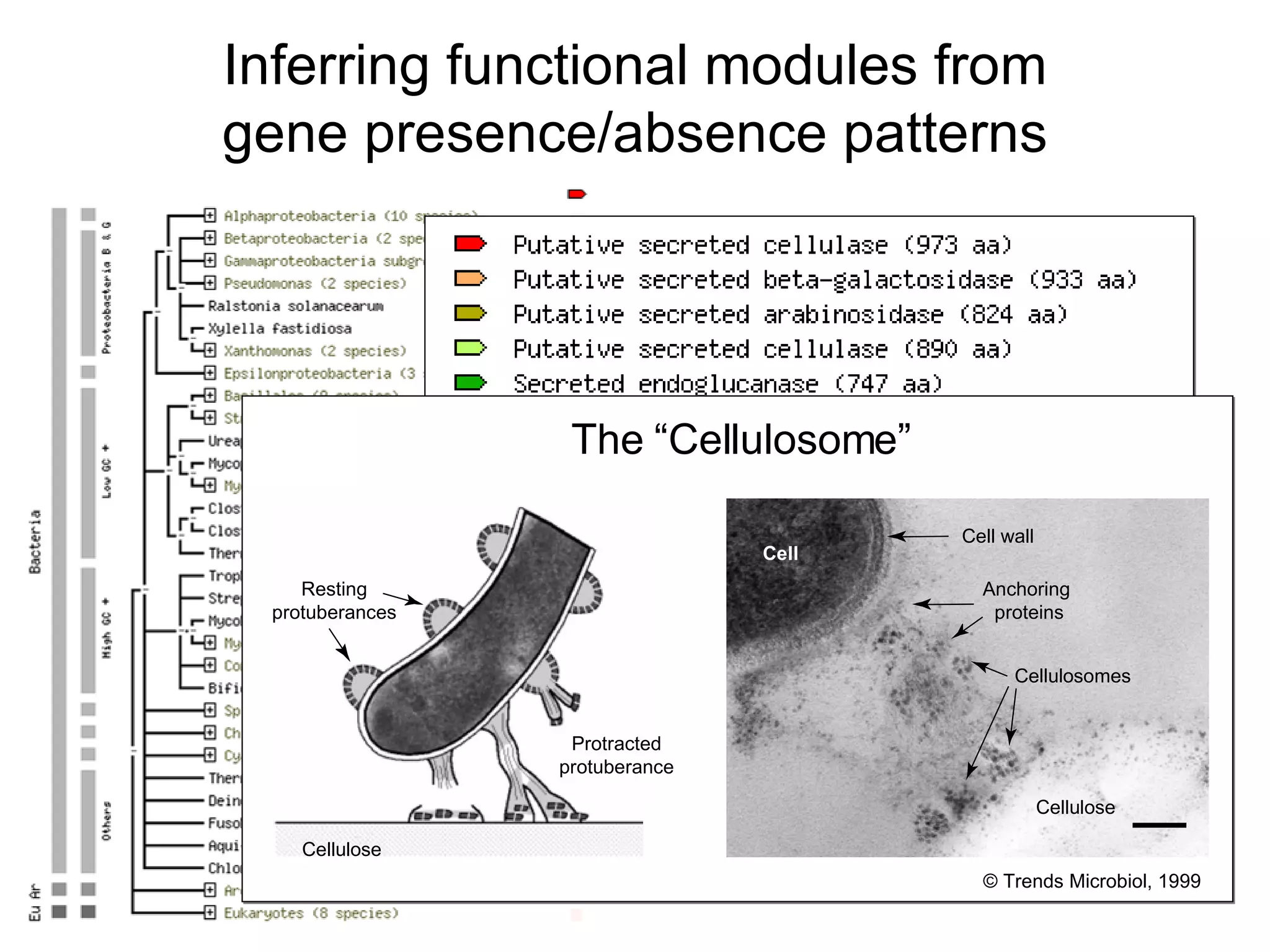

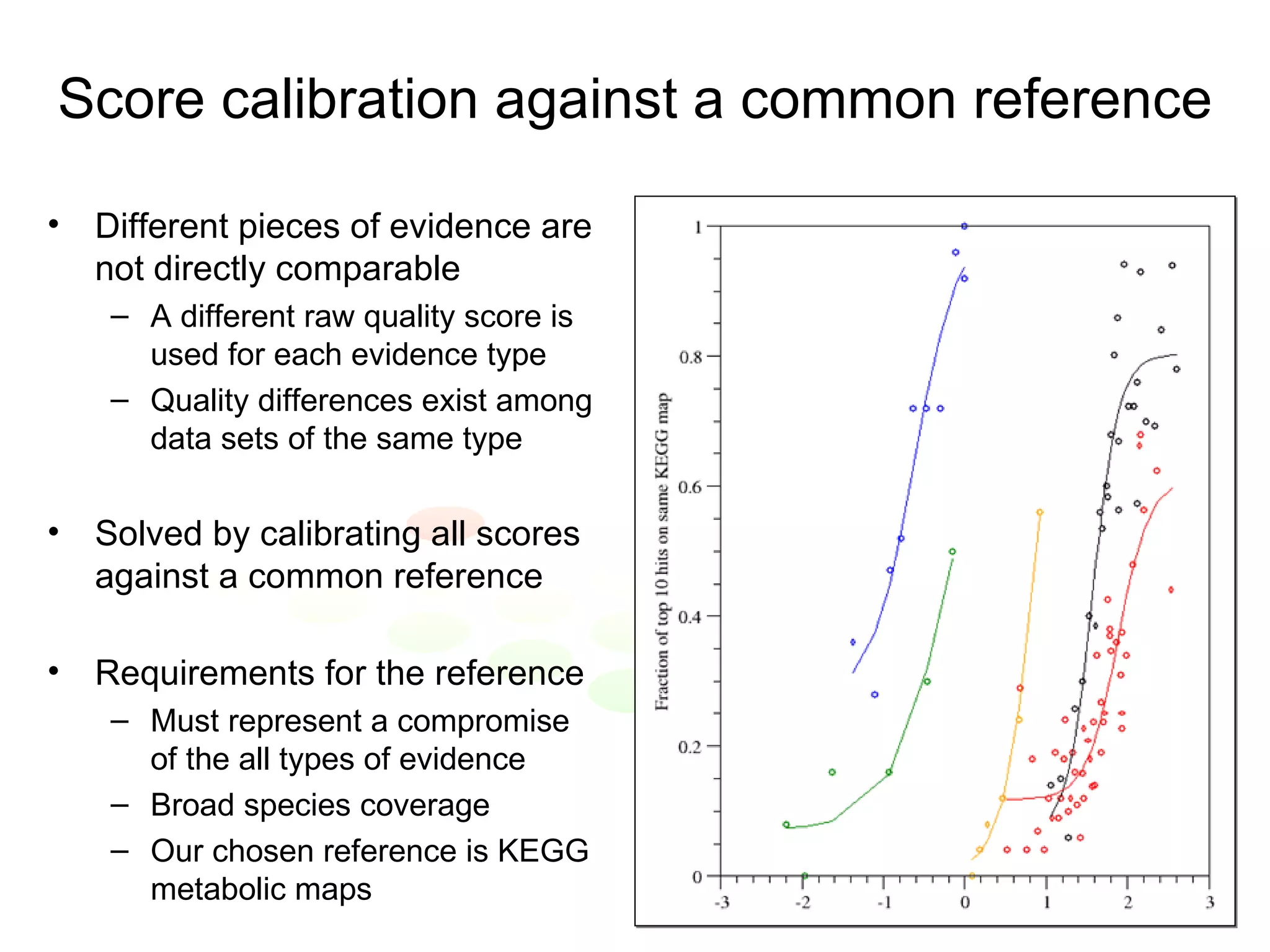
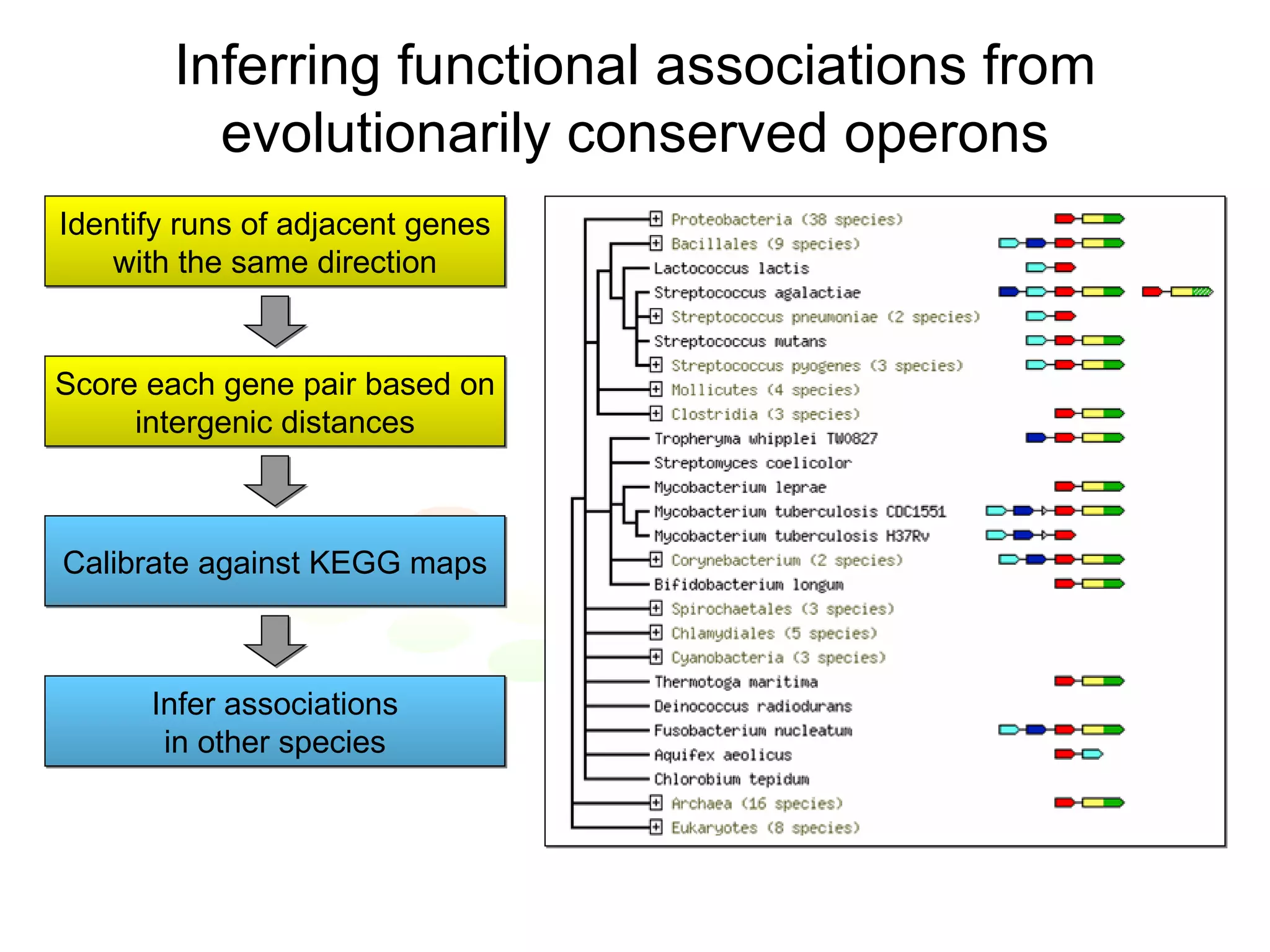
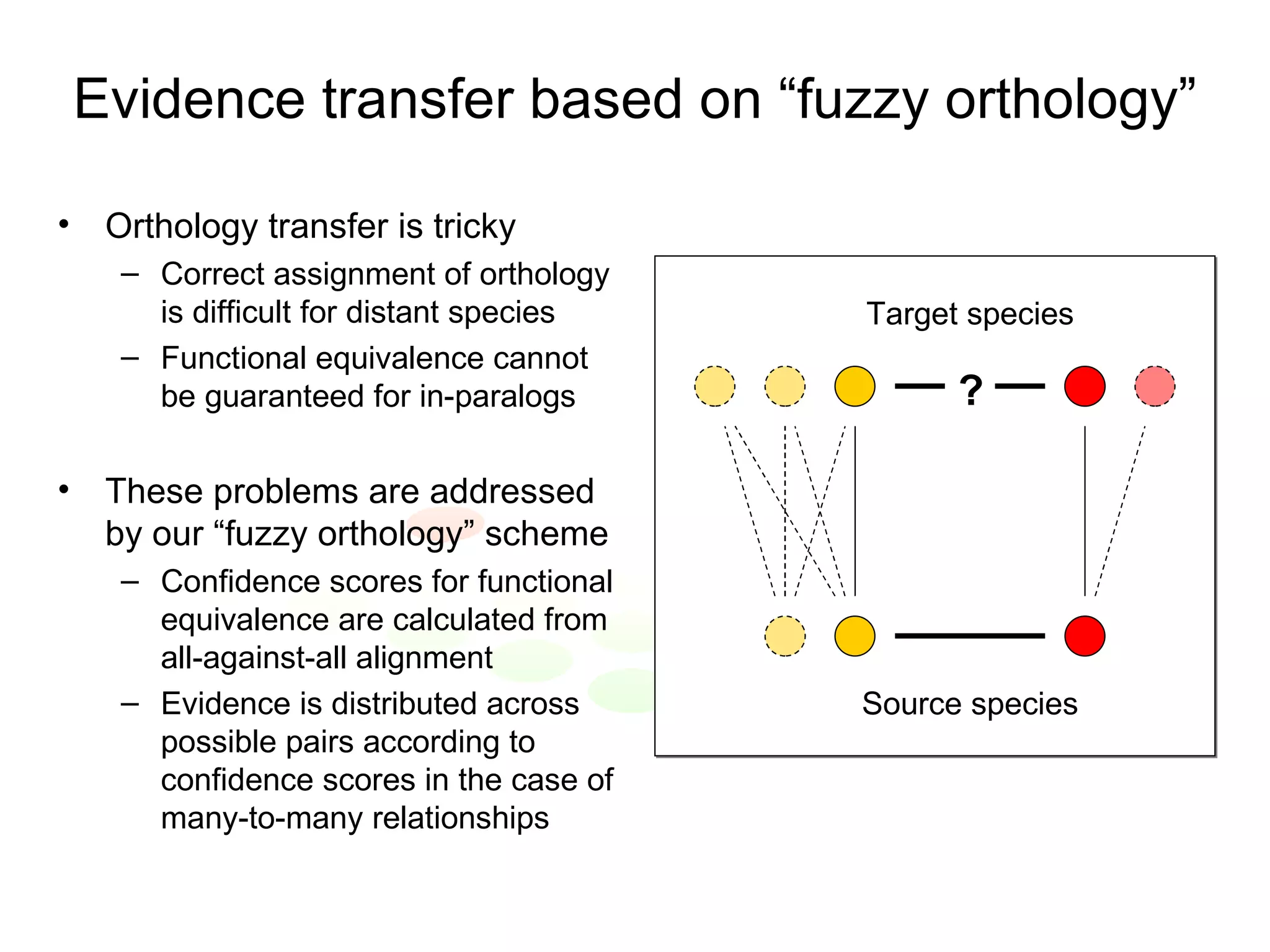
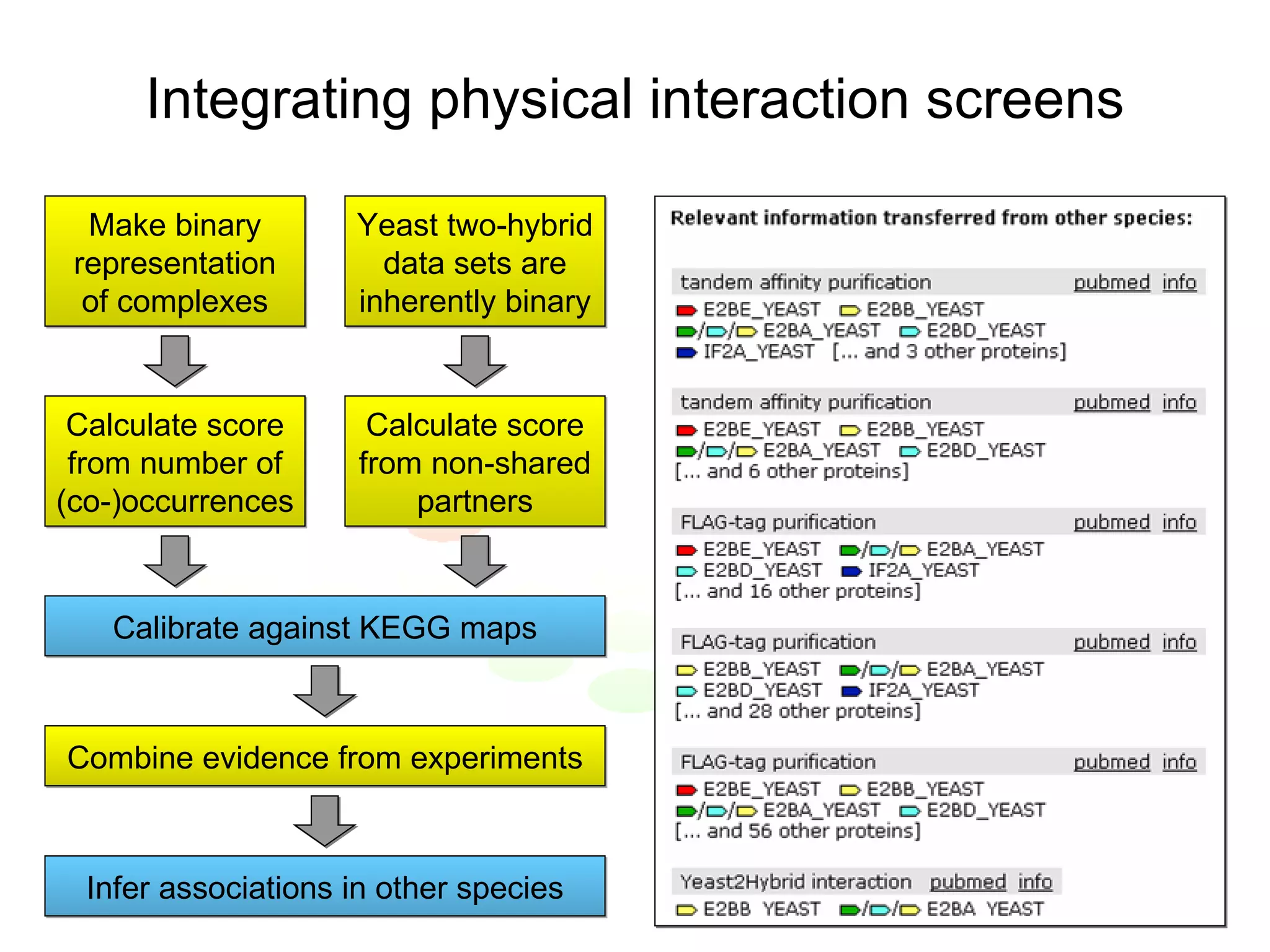
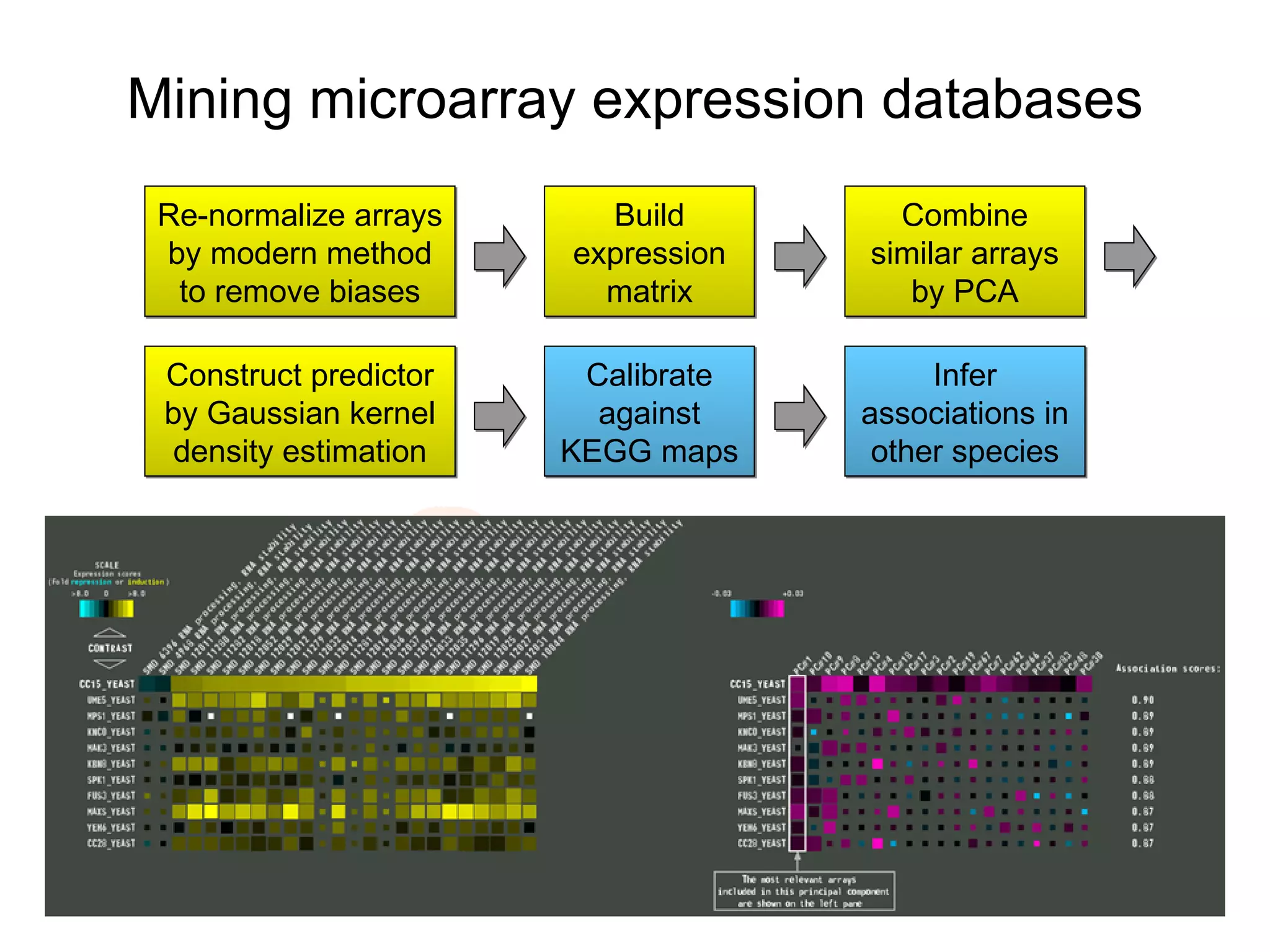
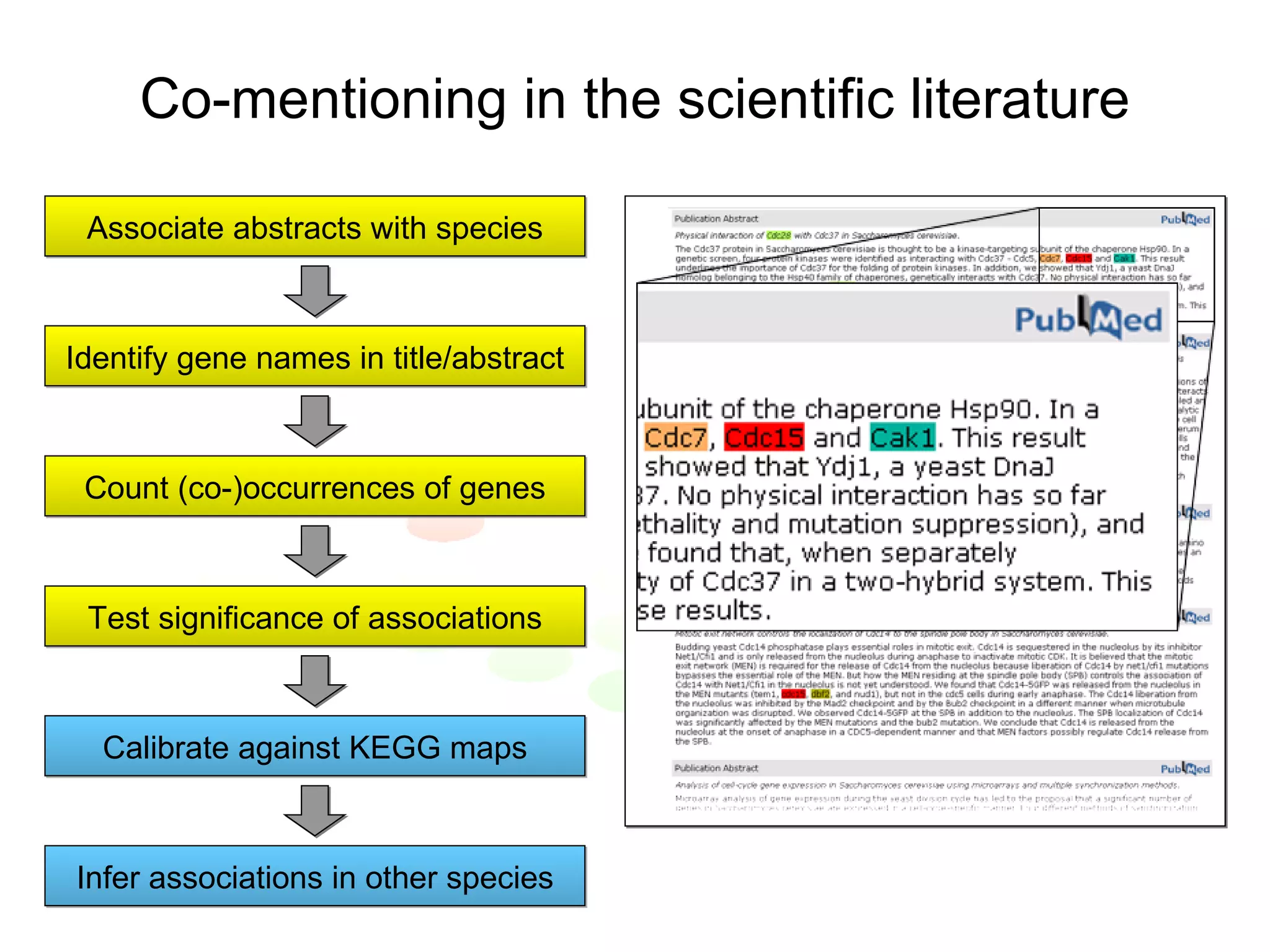
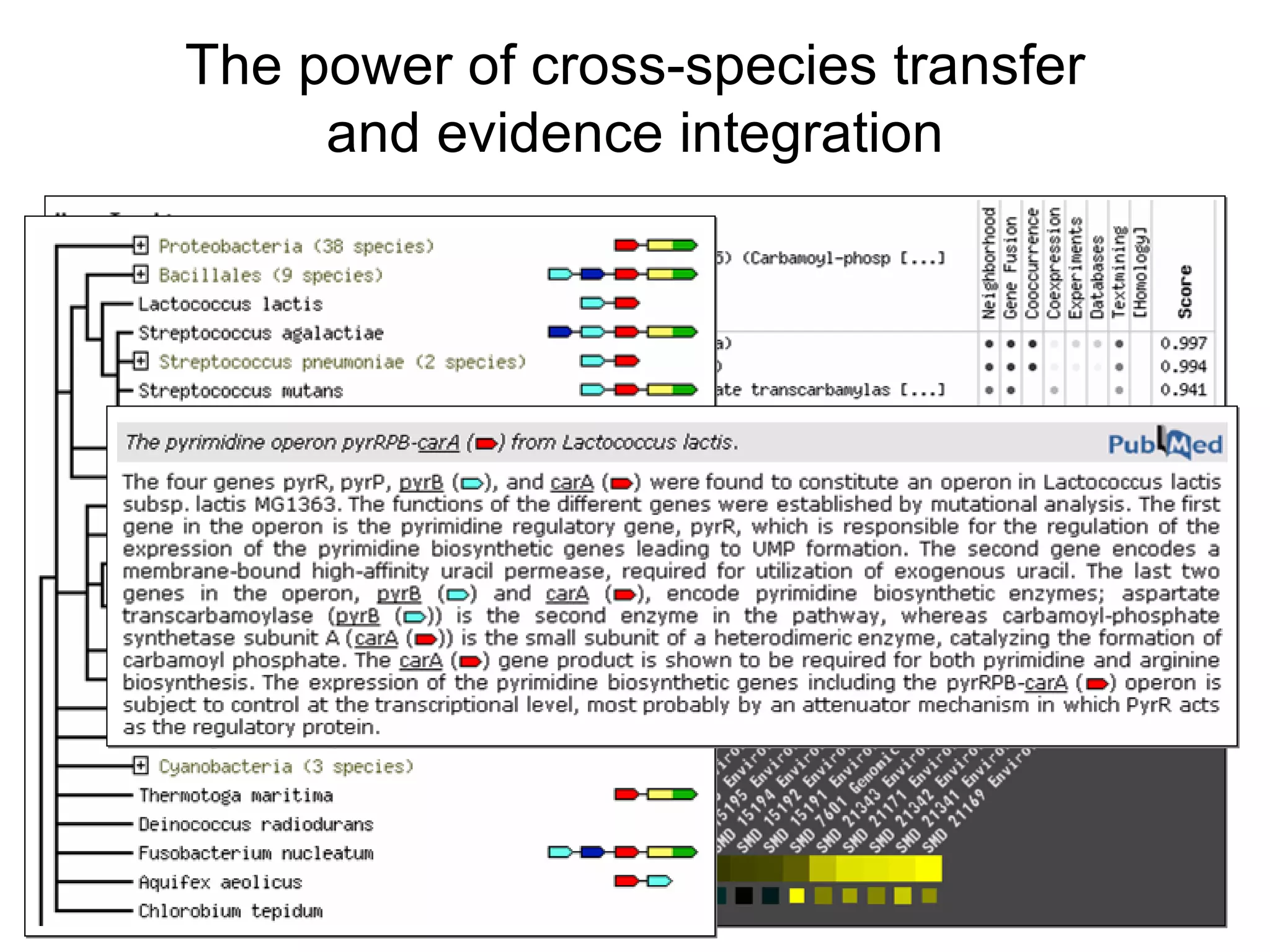
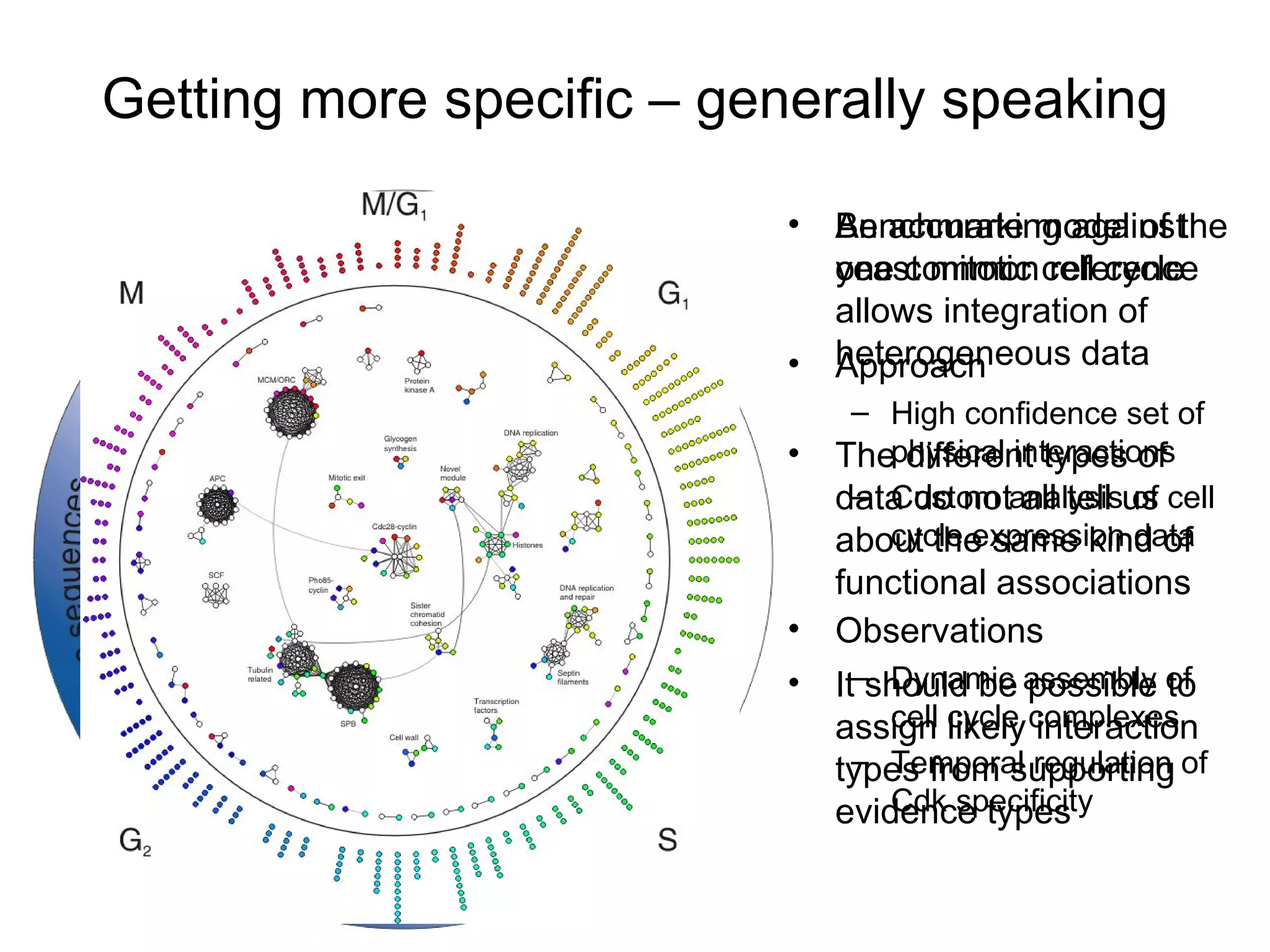




The document discusses the STRING web resource that integrates diverse large-scale genomic data across over 100 species, highlighting the challenges of data heterogeneity, standardization, and biases. It emphasizes the importance of high-quality data integration for predicting functional associations among proteins and proposes methods for inferring relationships through various evidence types. The update to the STRING database aims to enhance cross-species evidence transfer and improve the accuracy of data interpretation.














































































![Vibe Coding vs. Spec-Driven Development [Free Meetup]](https://cdn.slidesharecdn.com/ss_thumbnails/vibecodingvsspecdrivendevelopment-251209105622-43f455e7-thumbnail.jpg?width=640&height=640&fit=bounds)













