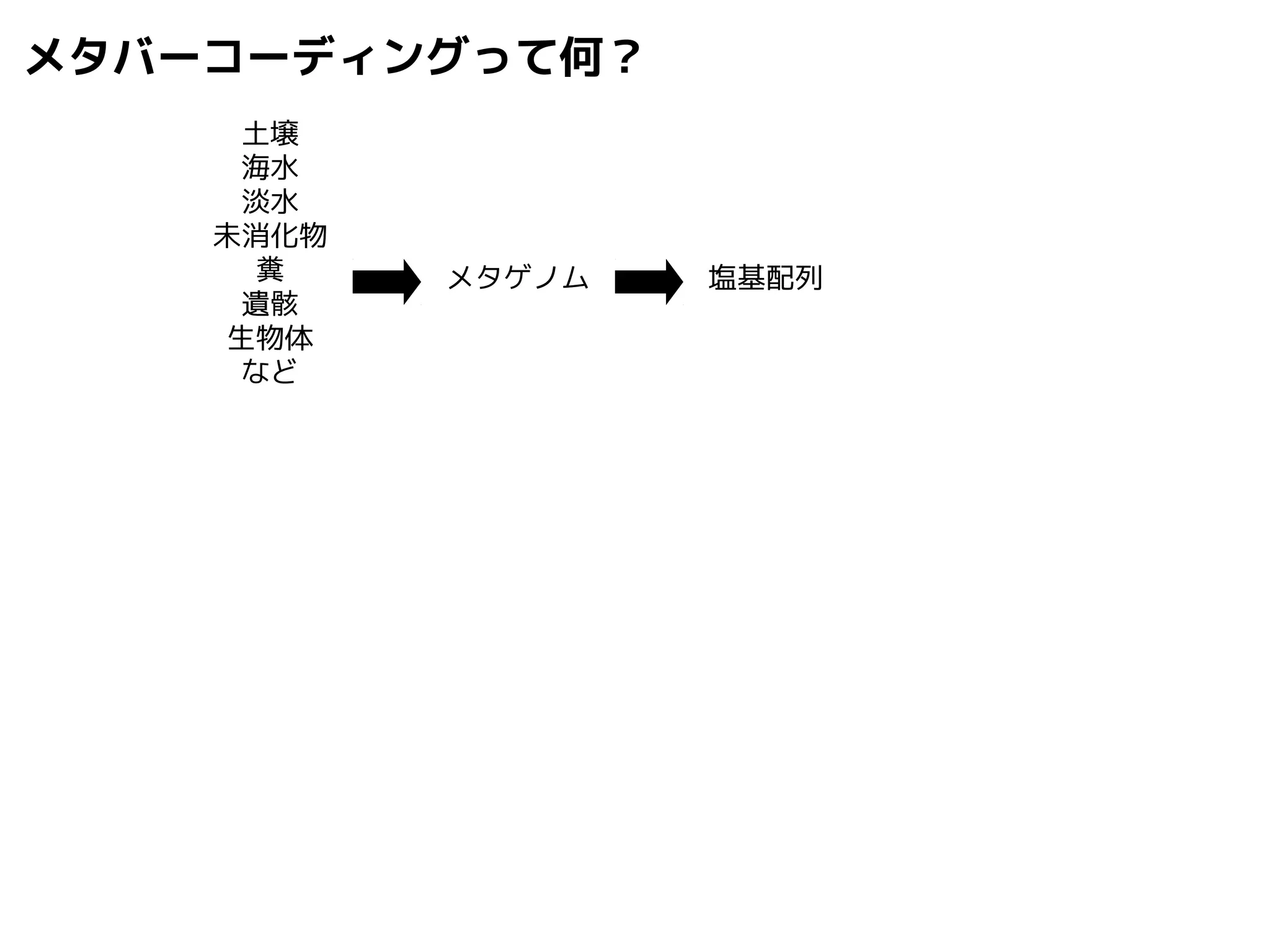
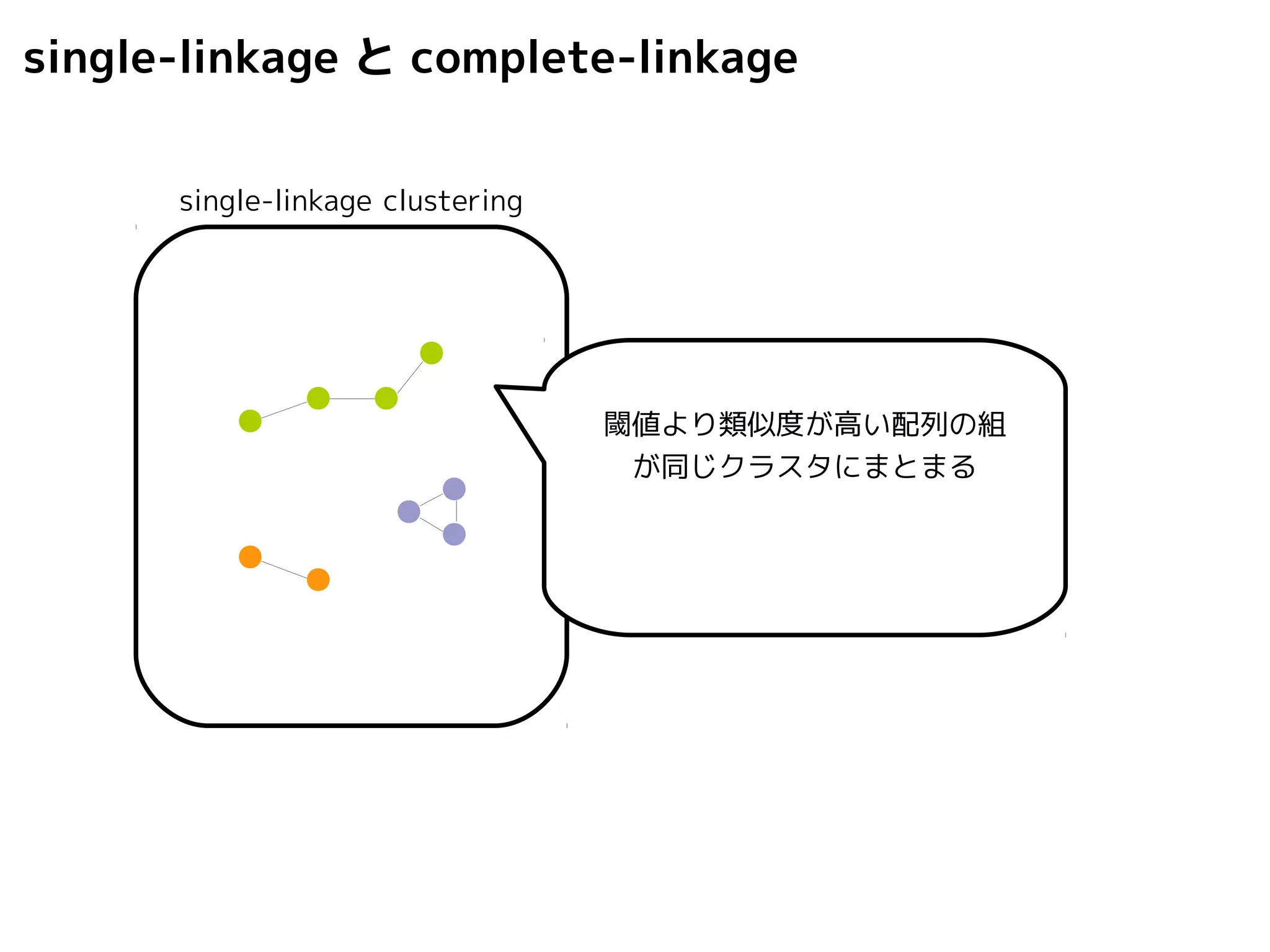
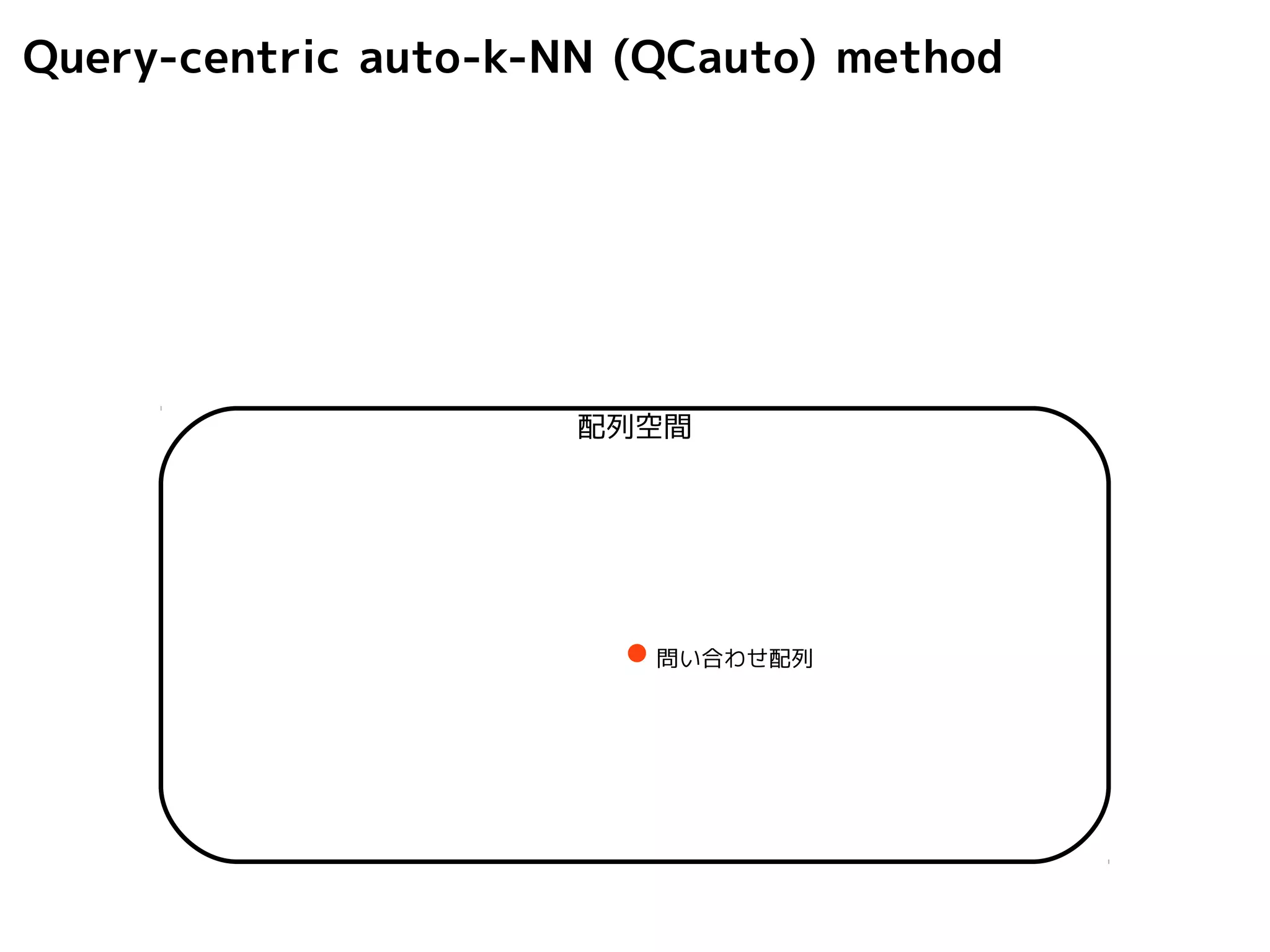
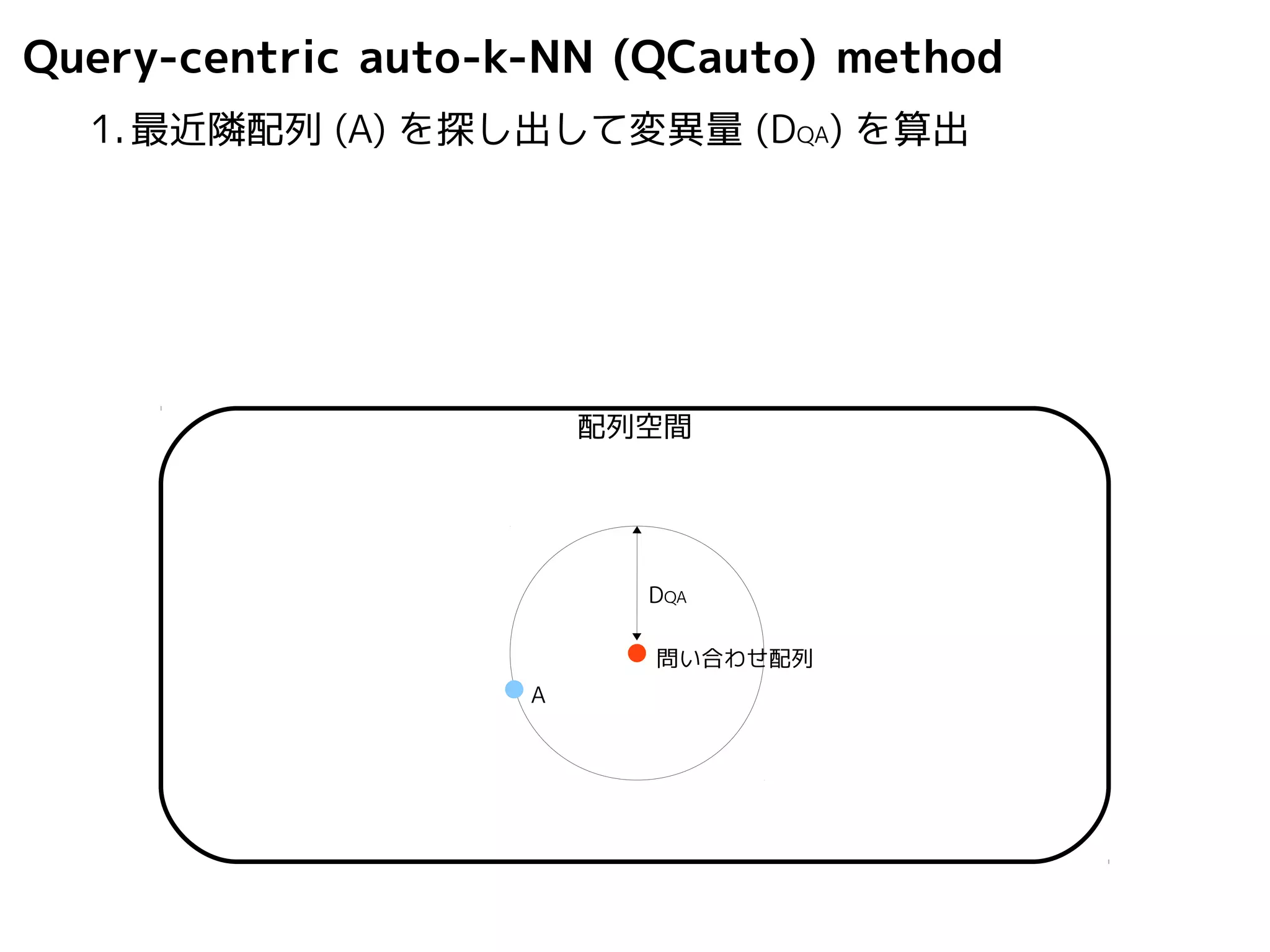
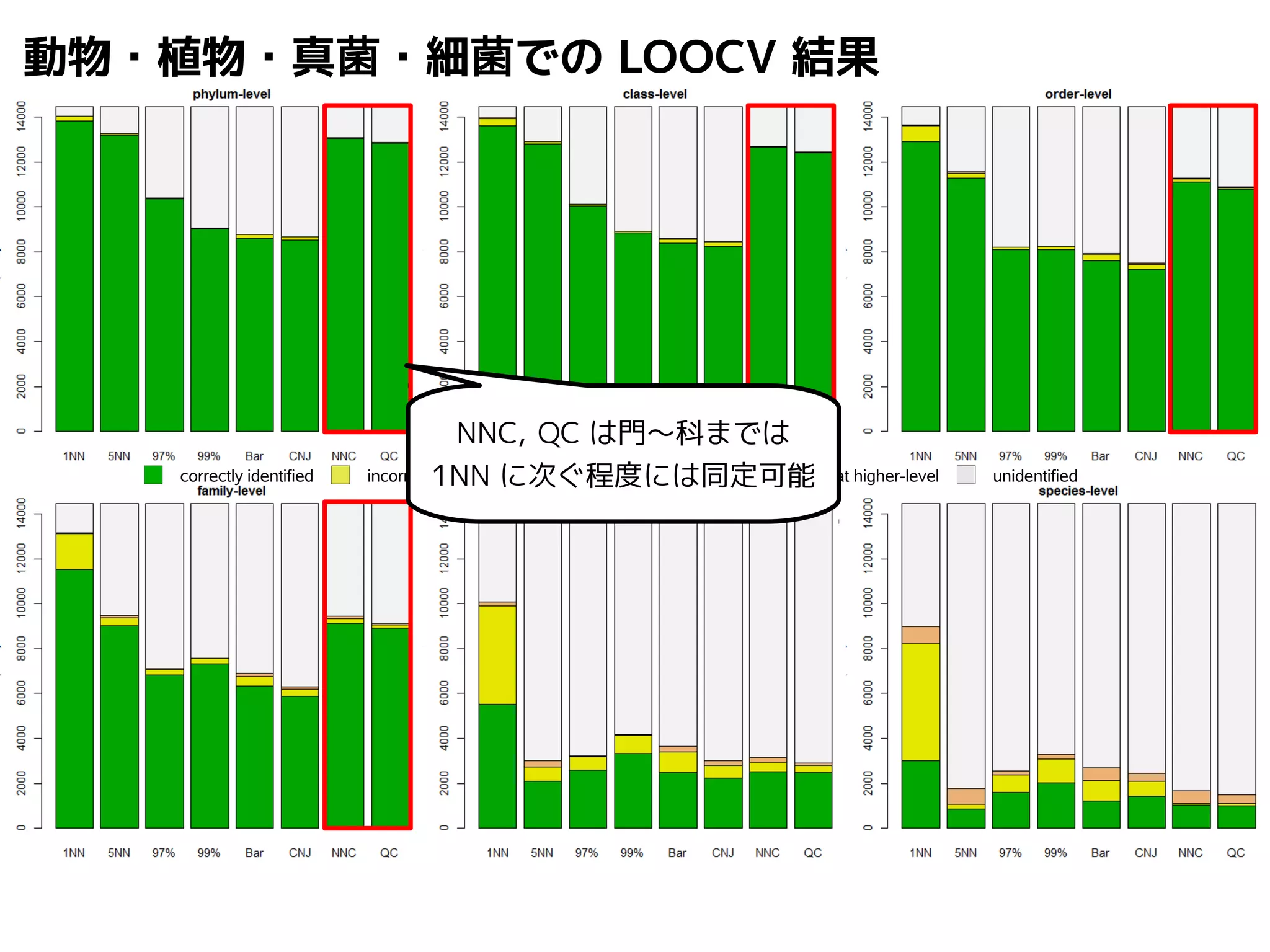
Query-centric auto-k-NN (QCauto)method
1.最近隣配列(A) を探し出して変異量(DQA) を算出
2.DAB>DQAを満たす配列のうち最もA に近い配列(B) を得る
3.DQN≤DQBを満たすすべての配列(N) を得る
A
DQB
N
N
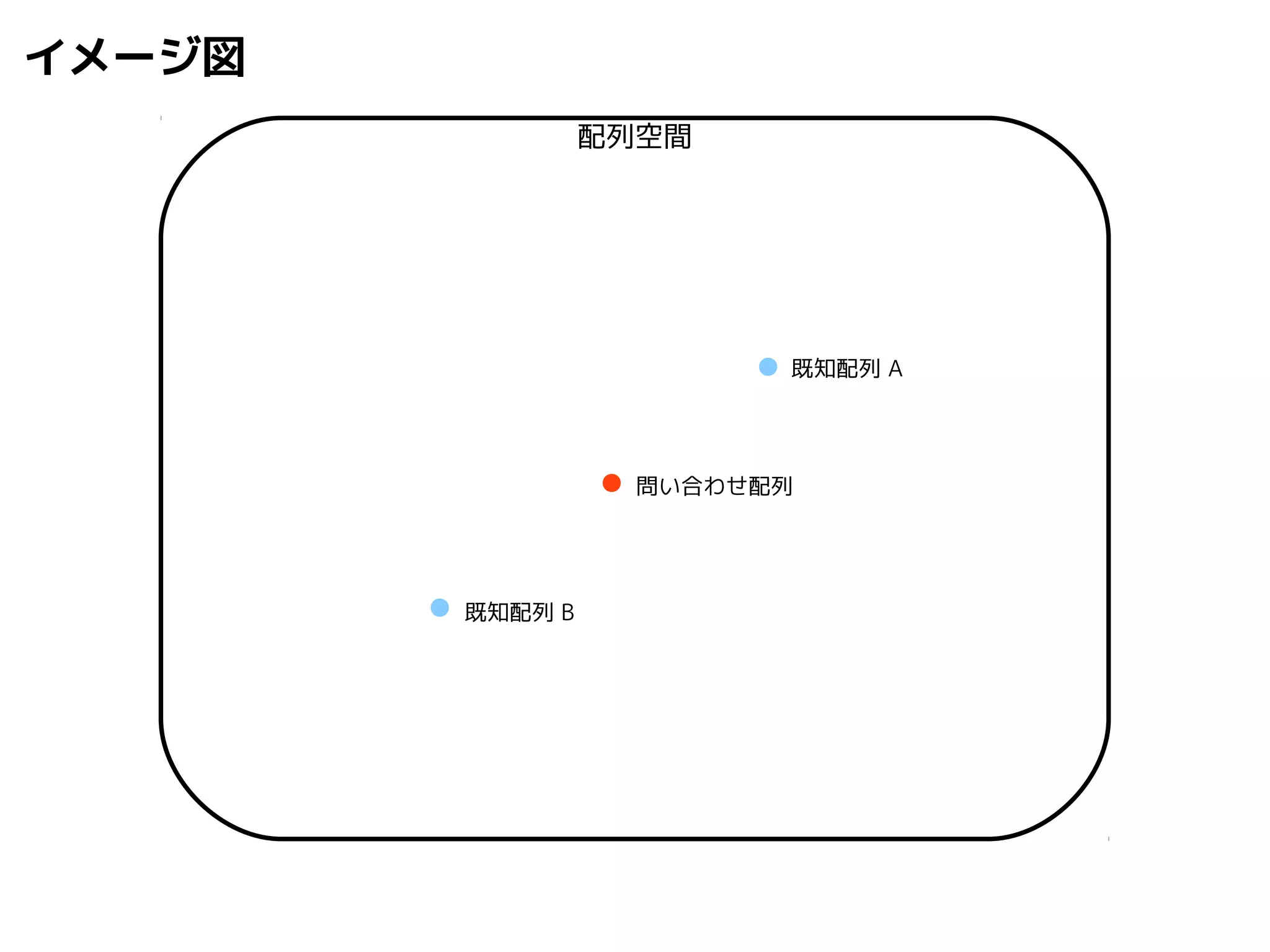
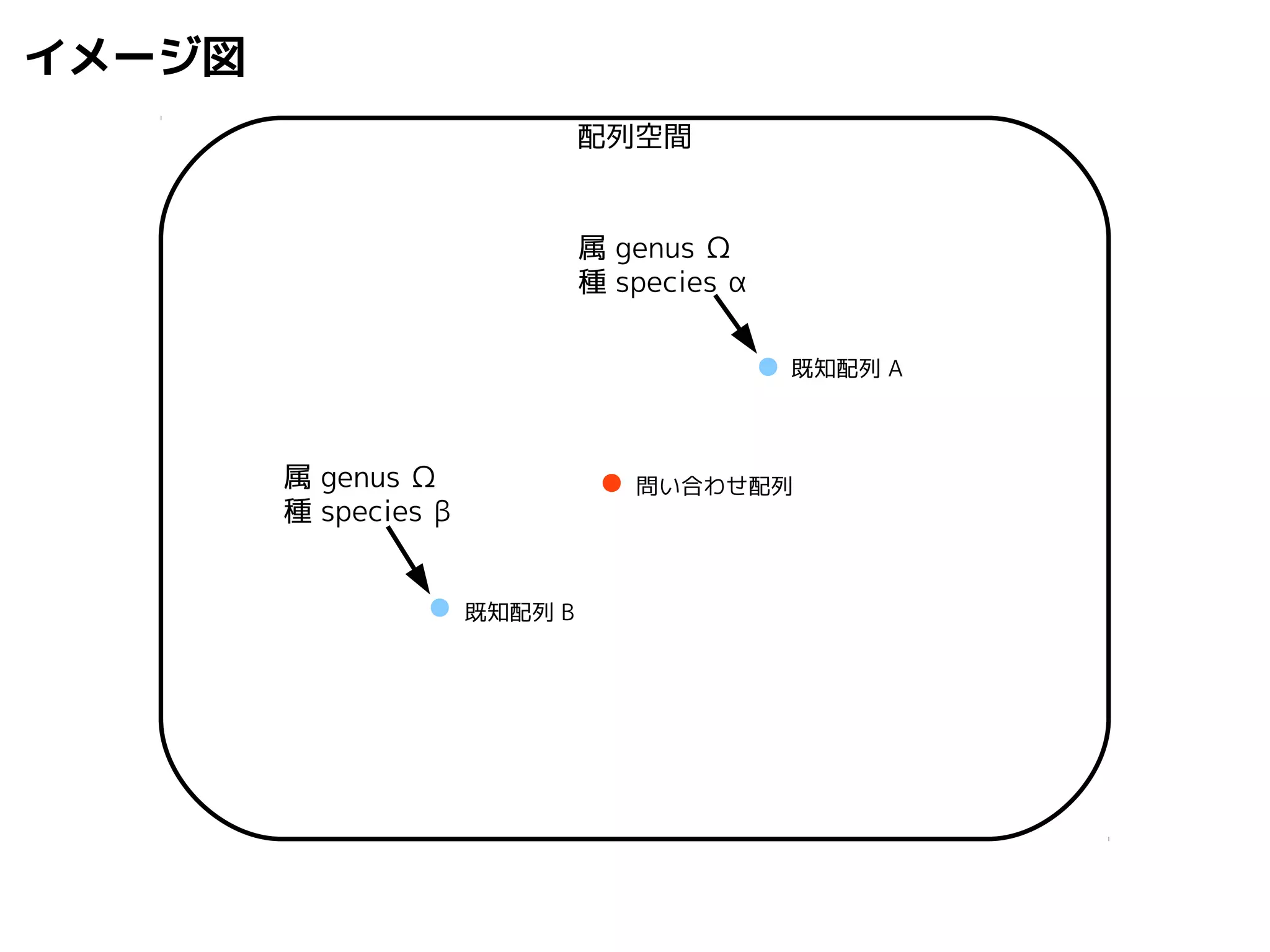
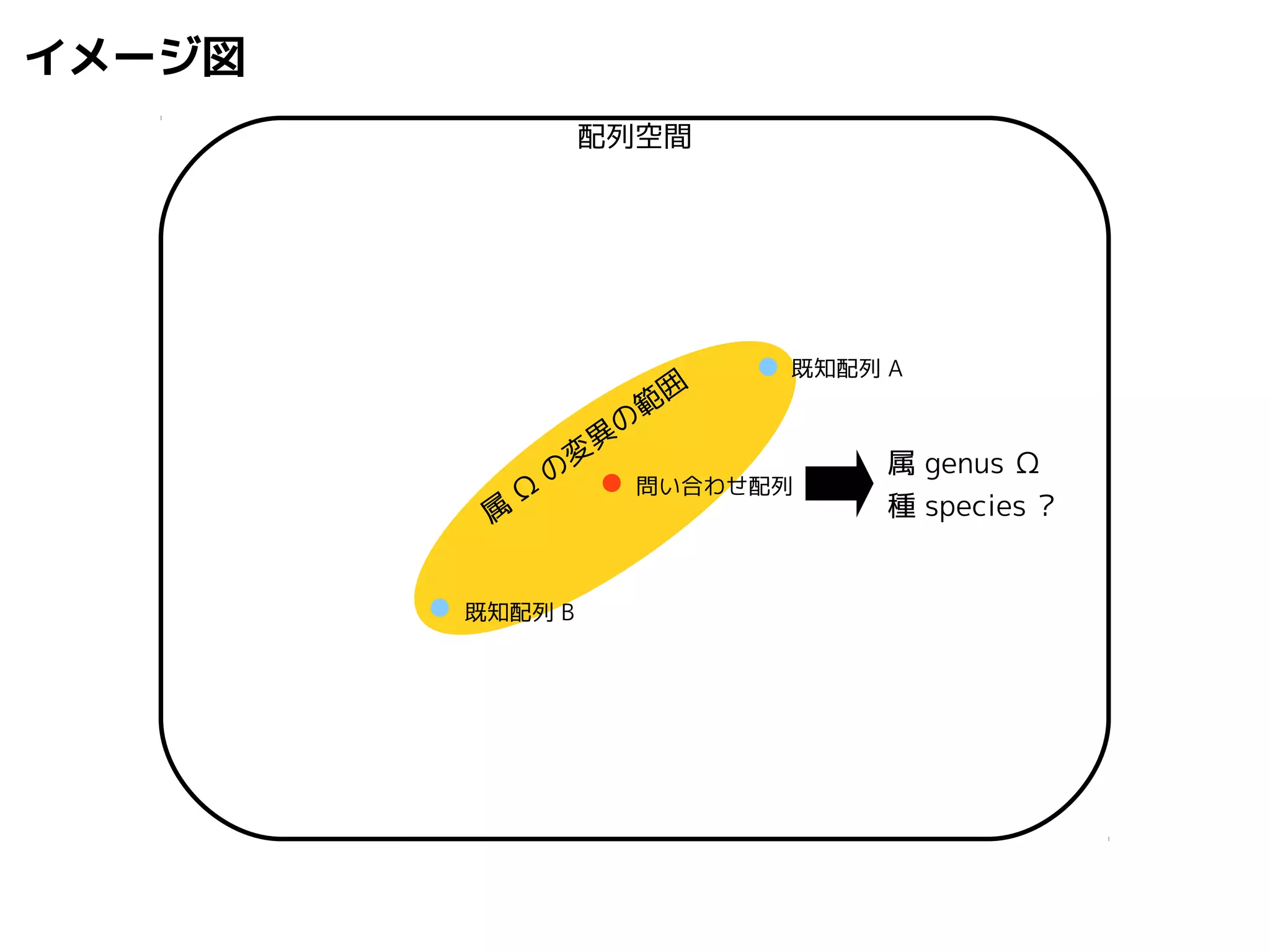
問い合わせ配列
N
配列空間
B
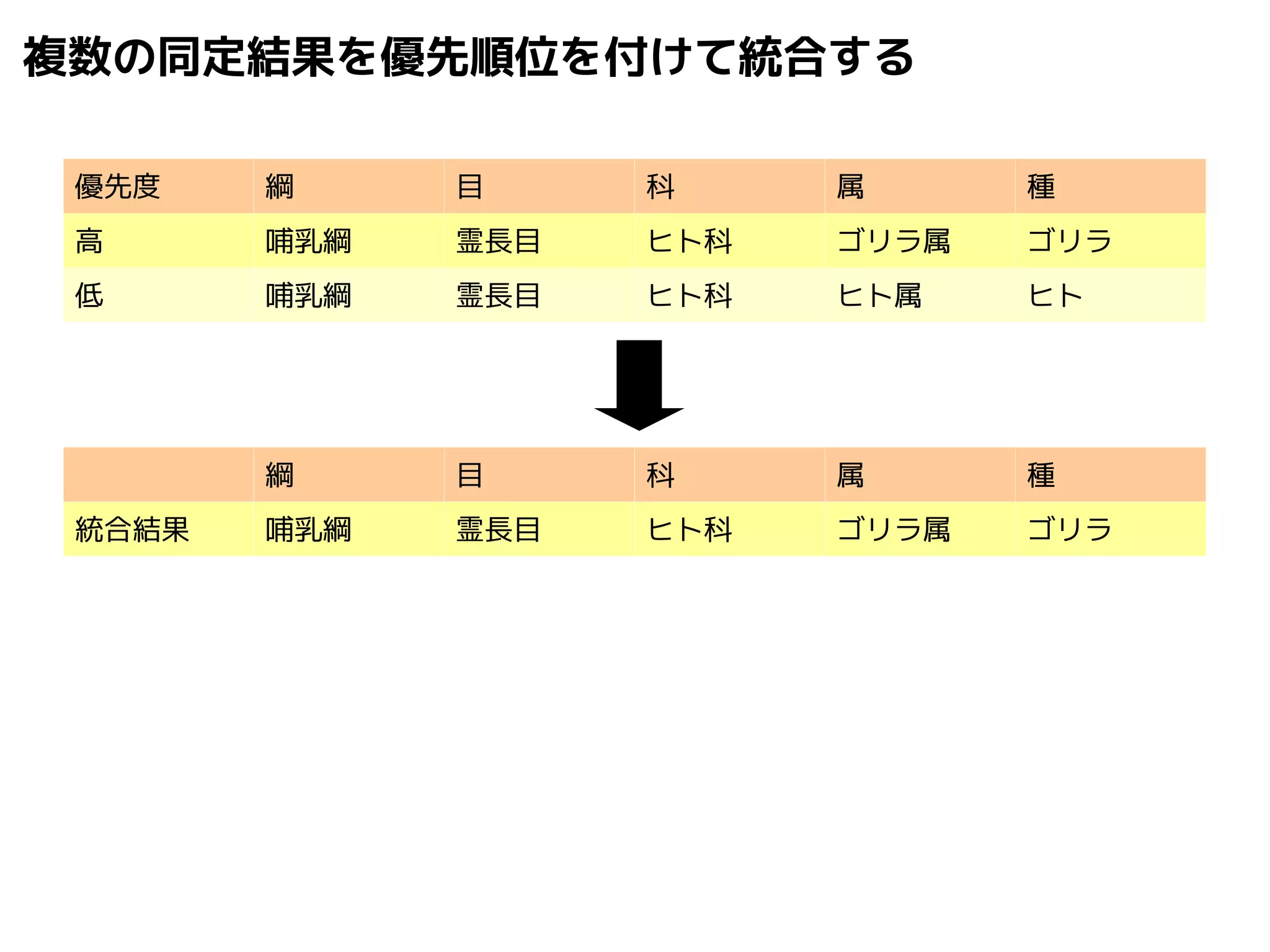
74.
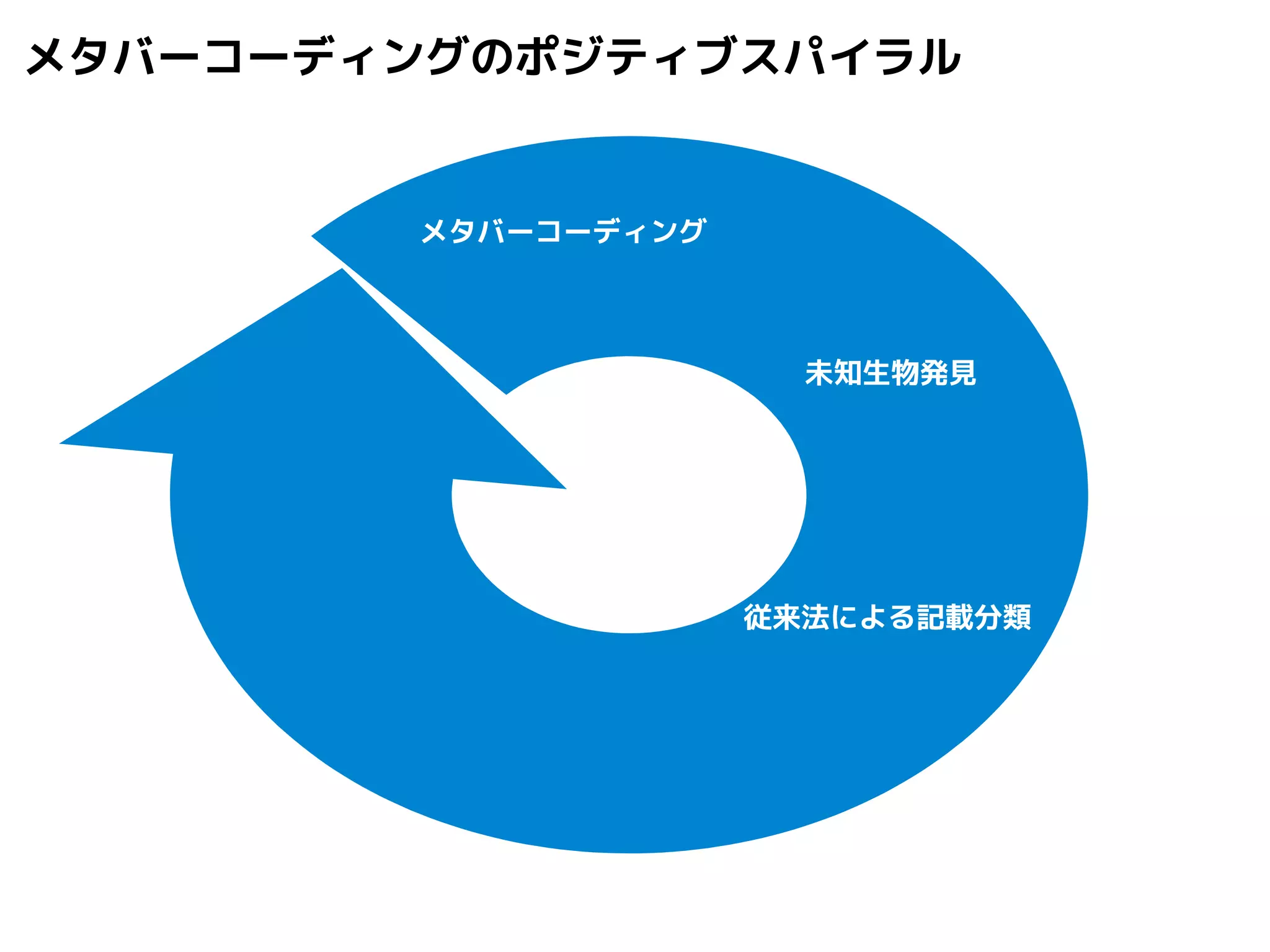
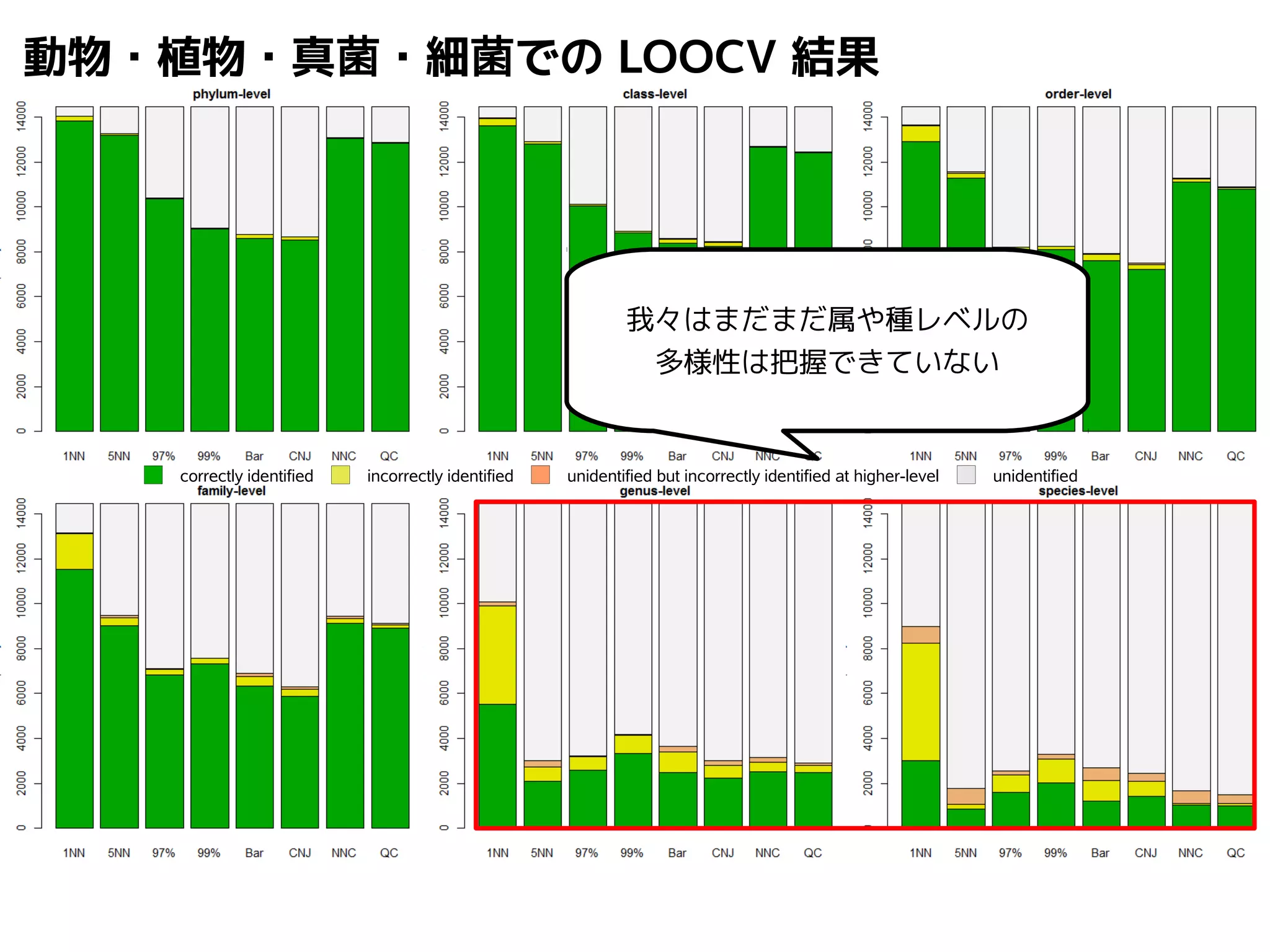
Query-centric auto-k-NN (QCauto)method
1.最近隣配列(A) を探し出して変異量(DQA) を算出
2.DAB>DQAを満たす配列のうち最もA に近い配列(B) を得る
3.DQN≤DQBを満たすすべての配列(N) を得る
4.A, B, N の全配列で共通する分類群を採用
A
DQB
N
N
問い合わせ配列
N
配列空間
B
75.
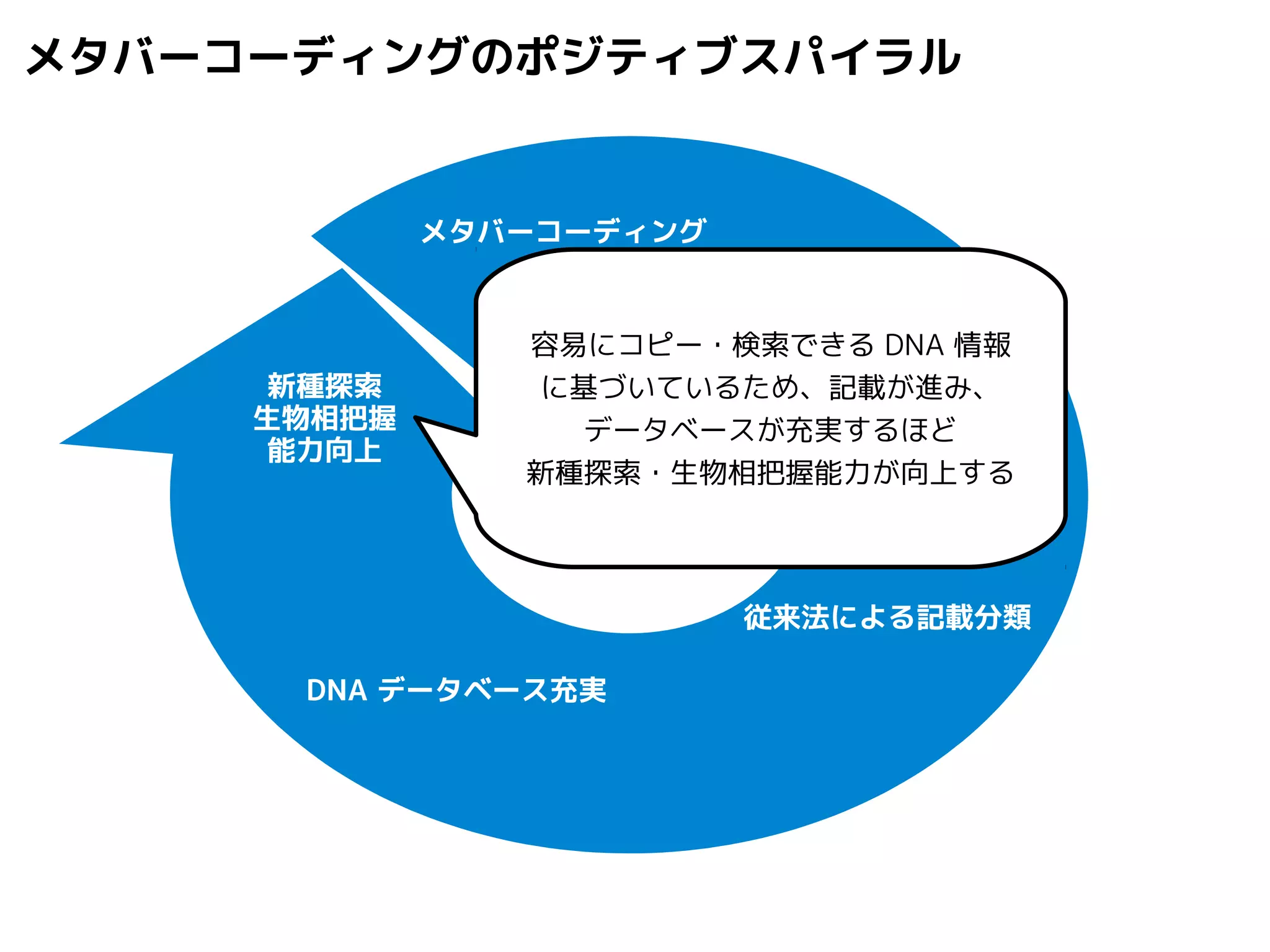
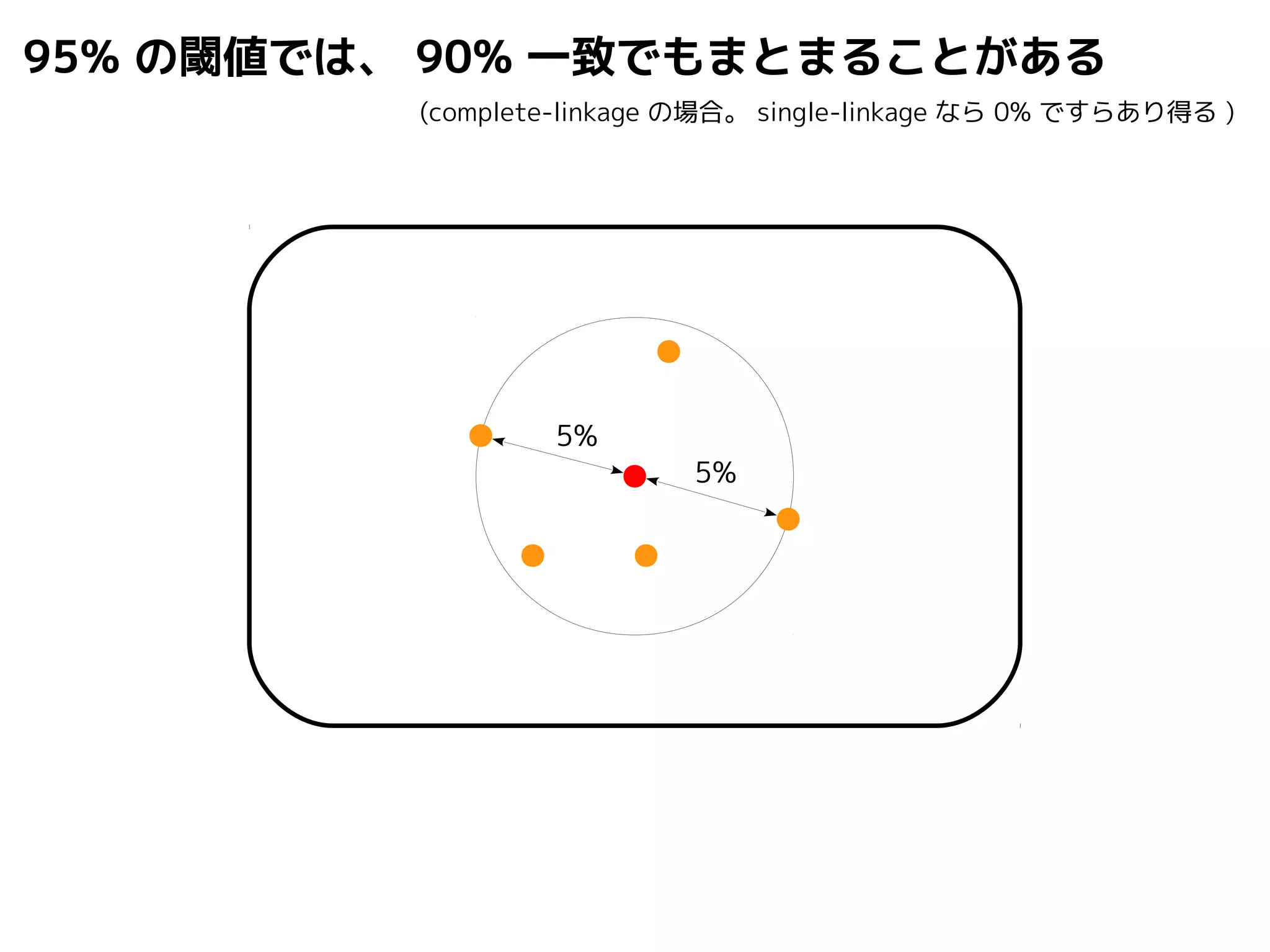
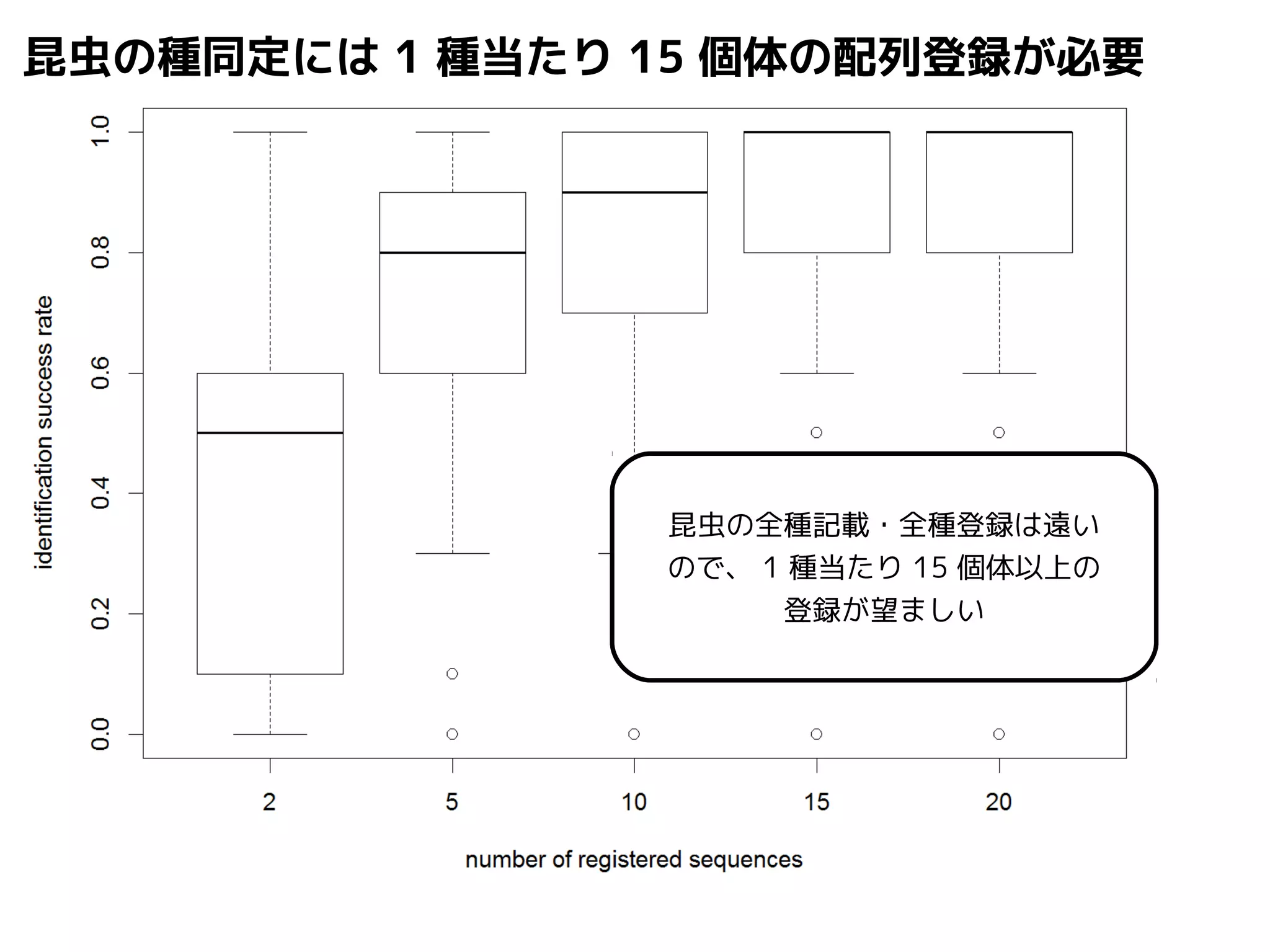
Query-centric auto-k-NN (QCauto)method
1.最近隣配列(A) を探し出して変異量(DQA) を算出
問い合わせ配列と最近隣配列間の変異量
2.DAB>DQAを満たす配列のうち最もA に近い配列(B) を得る
3.DQN≤DQBを満たすすべての配列(N) を得る
4.A, B, N の全配列で共通する分類群を採用
< ≤ =
A
DQB
N
N
問い合わせ配列
N
配列空間
B
DQA
DQB
同定結果分類群内の最大変異量
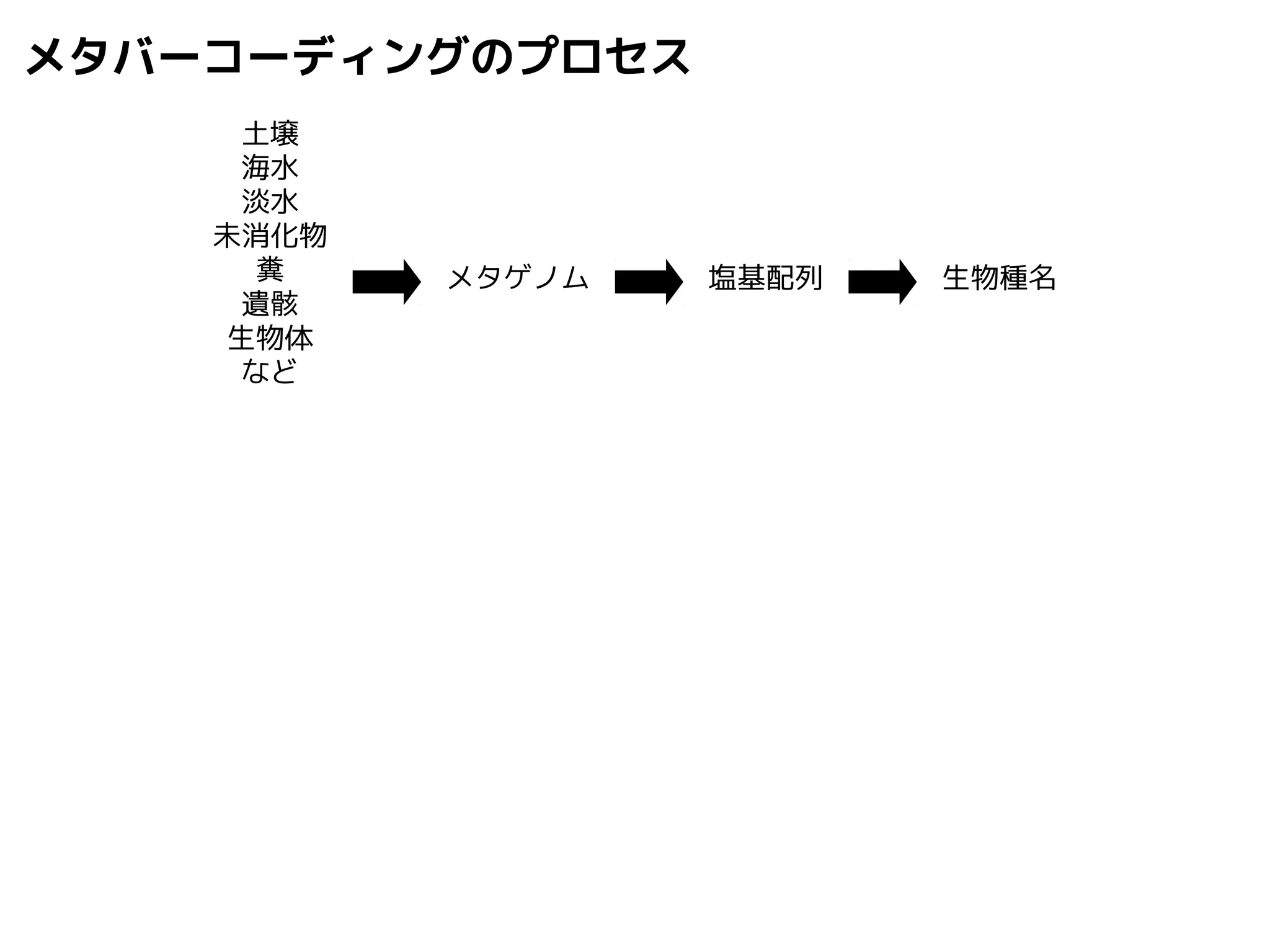
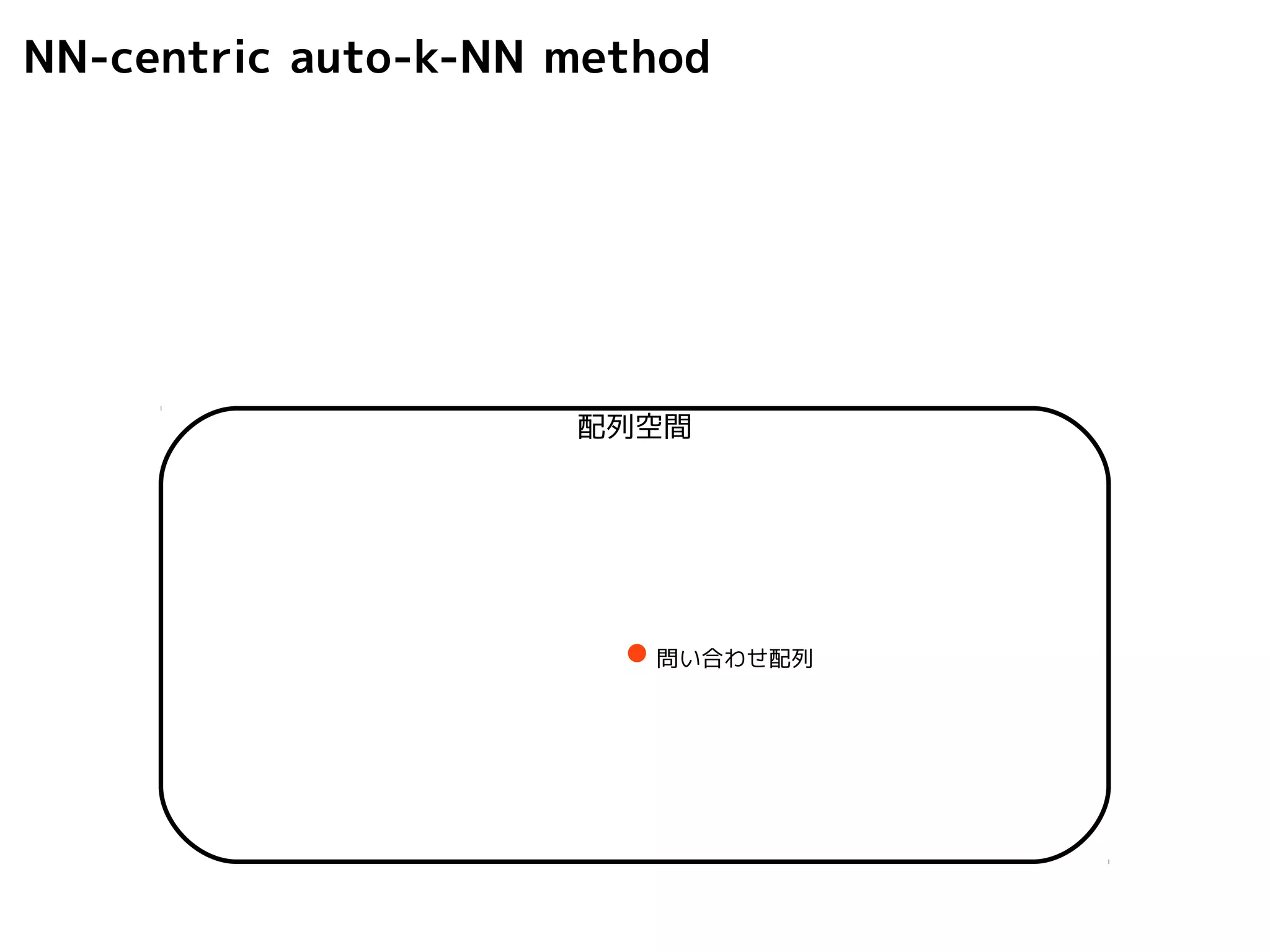
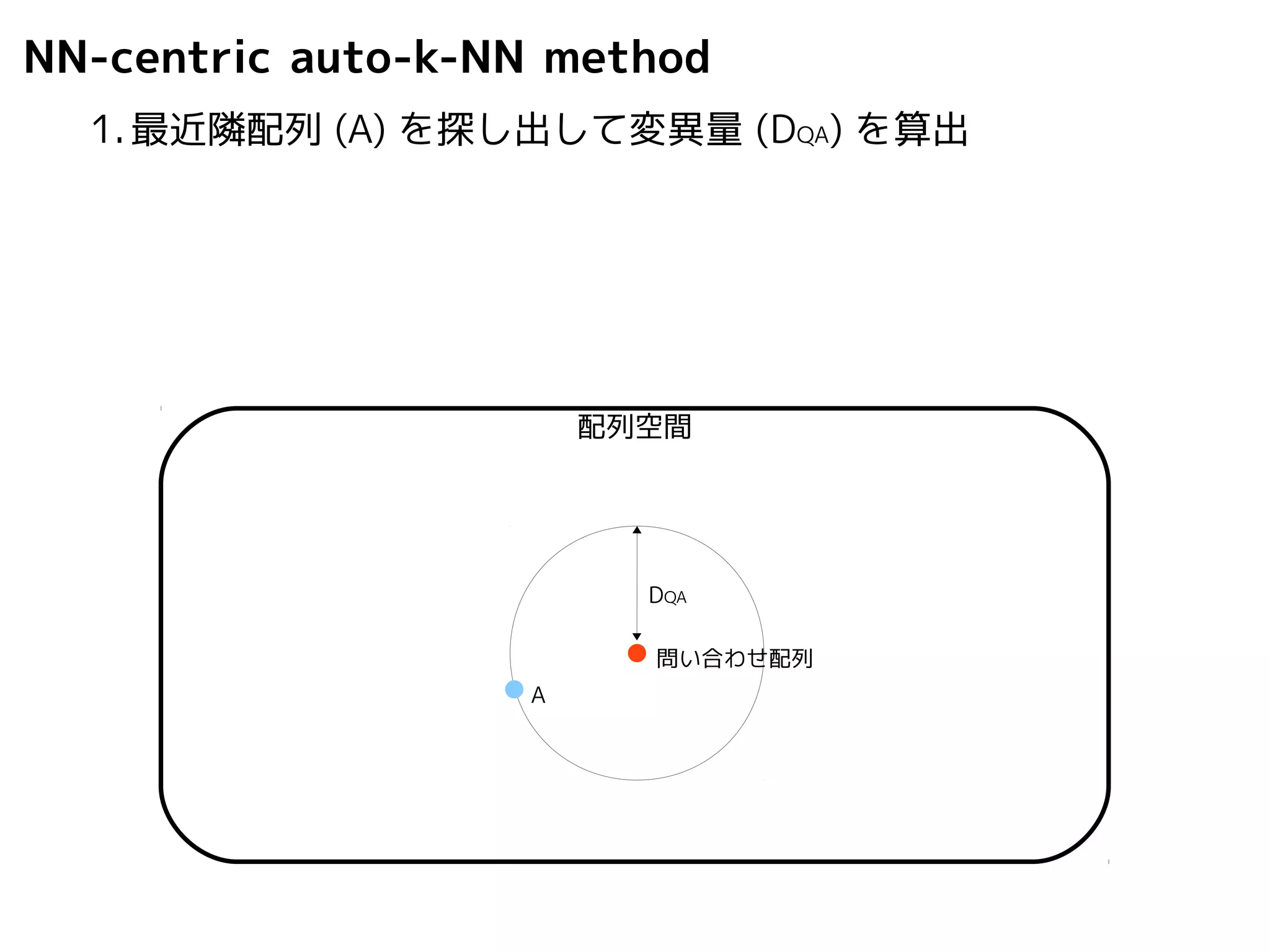
NN-centric auto-k-NN method
1.最近隣配列(A) を探し出して変異量(DQA) を算出
2.DAB>DQAを満たす配列のうち最もA に近い配列(B) を得る
DQA
A
配列空間
問い合わせ配列
B
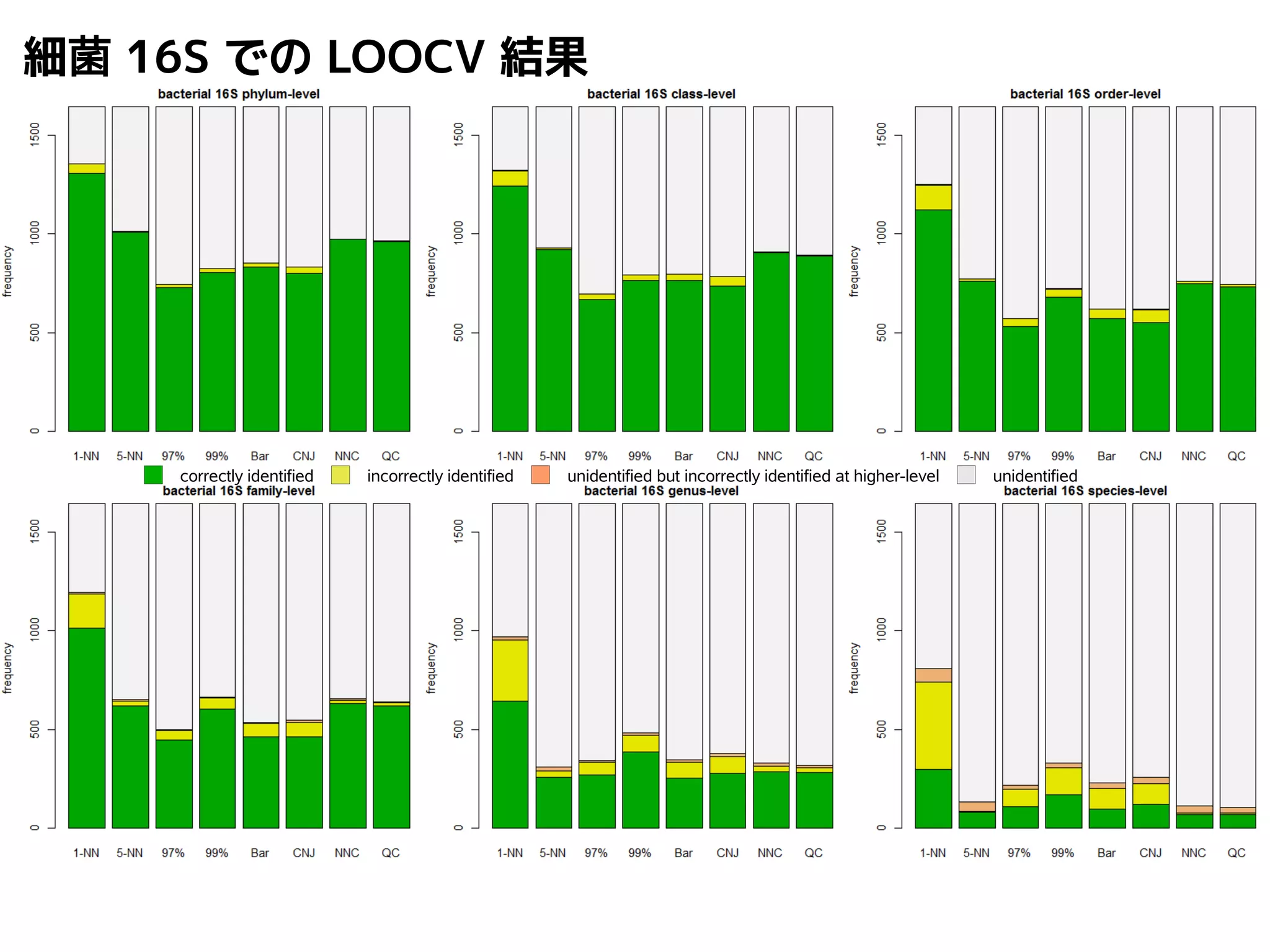
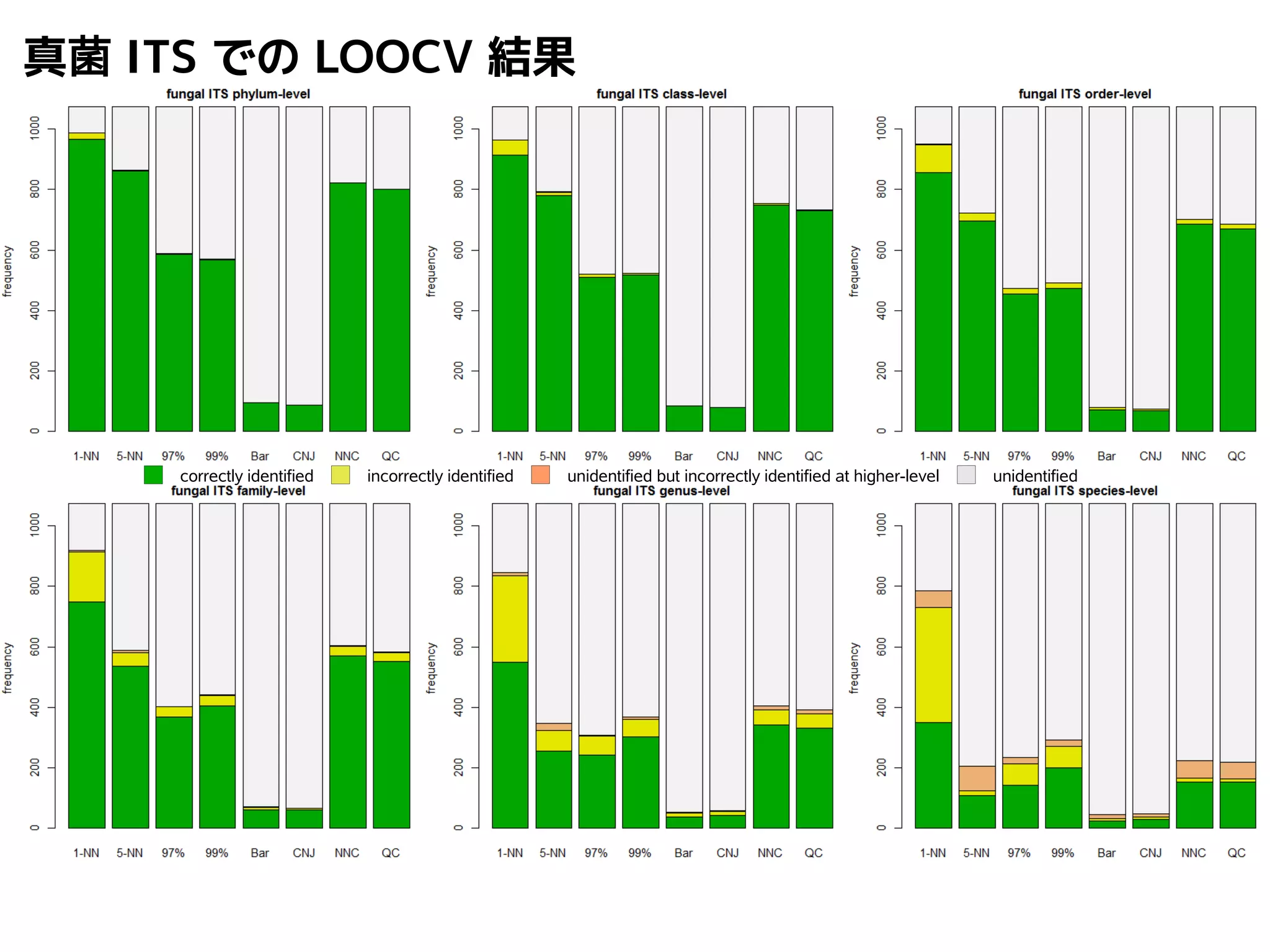
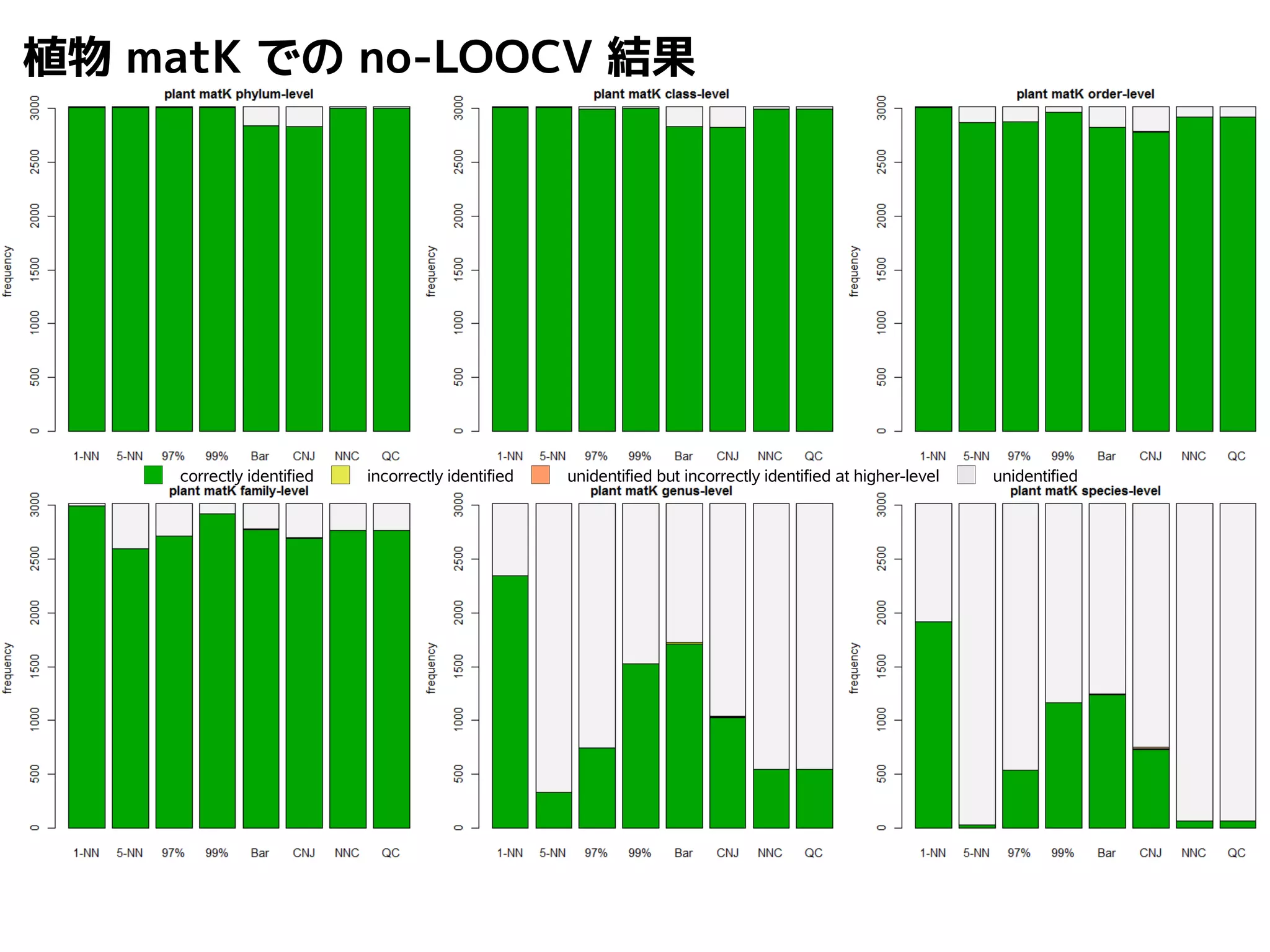
101.
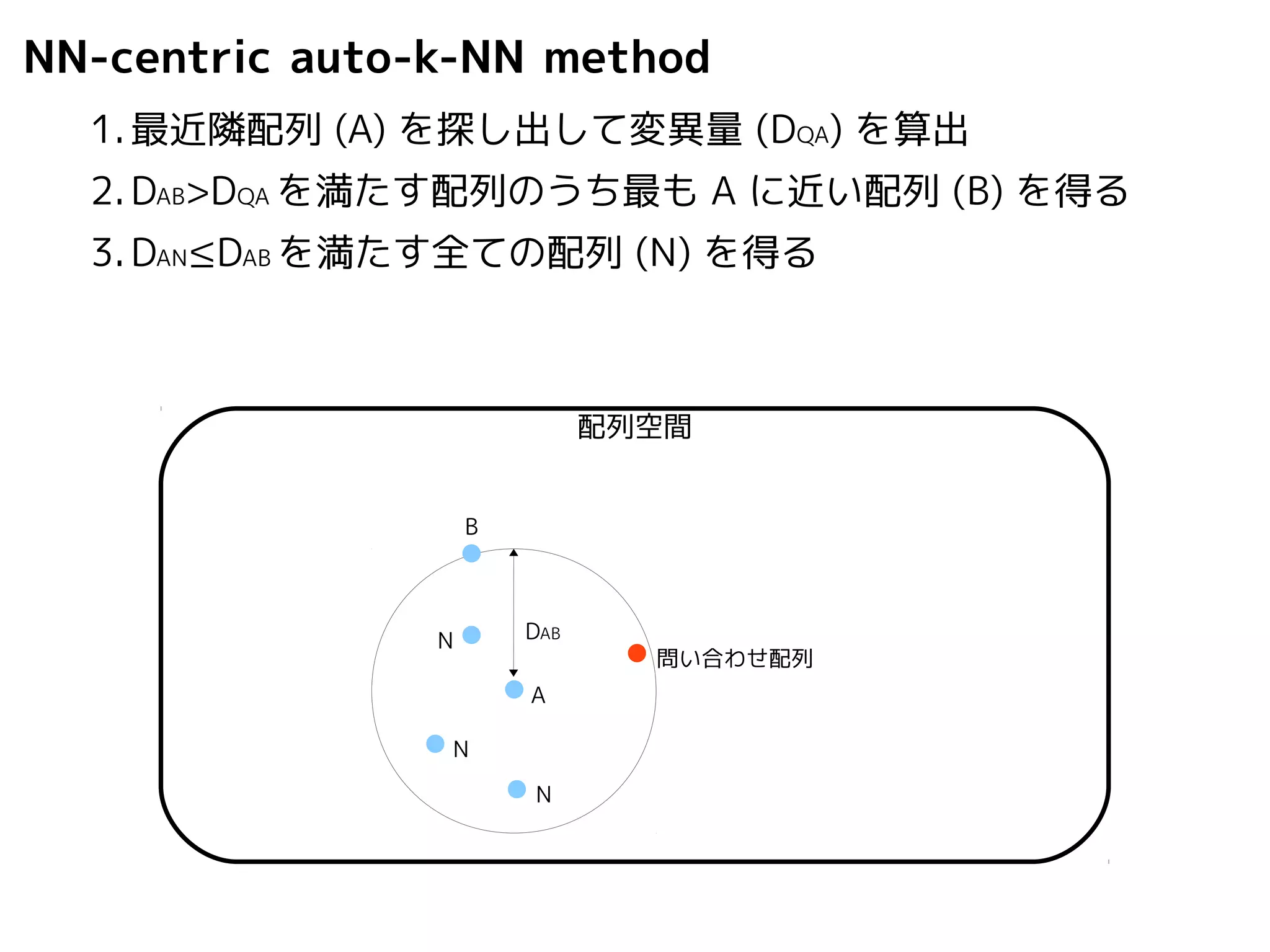
NN-centric auto-k-NN method
1.最近隣配列(A) を探し出して変異量(DQA) を算出
2.DAB>DQAを満たす配列のうち最もA に近い配列(B) を得る
3.DAN≤DABを満たす全ての配列(N) を得る
DAB
A
B
N
N
N
配列空間
問い合わせ配列
102.
NN-centric auto-k-NN method
1.最近隣配列(A) を探し出して変異量(DQA) を算出
2.DAB>DQAを満たす配列のうち最もA に近い配列(B) を得る
3.DAN≤DABを満たす全ての配列(N) を得る
4.A, B, N の全配列で共通する分類群を採用
DAB
A
B
N
N
N
配列空間
問い合わせ配列
103.
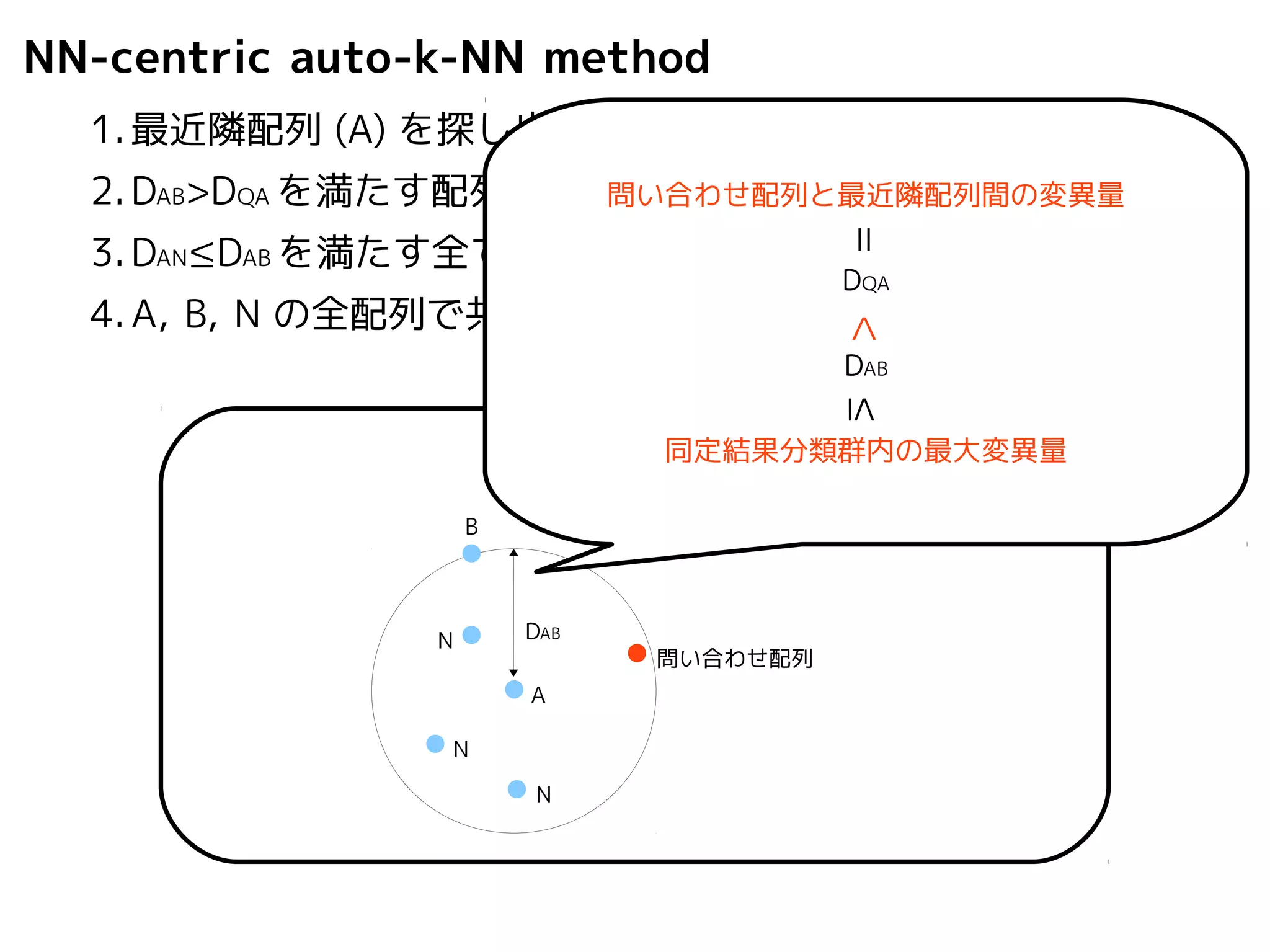
NN-centric auto-k-NN method
1.最近隣配列(A) を探し出して変異量(DQA) を算出
2.DAB>DQAを満たす配列のうち最もA に近い配列(B) を得る
3.DAN≤DABを満たす全ての配列(N) を得る
4.A, B, N の全配列で共通する分類群を採用
DAB
A
B
N
N
N
問い合わせ配列と最近隣配列間の変異量
配列空間
問い合わせ配列
=
DQA
< DAB
≤ 同定結果分類群内の最大変異量

















































































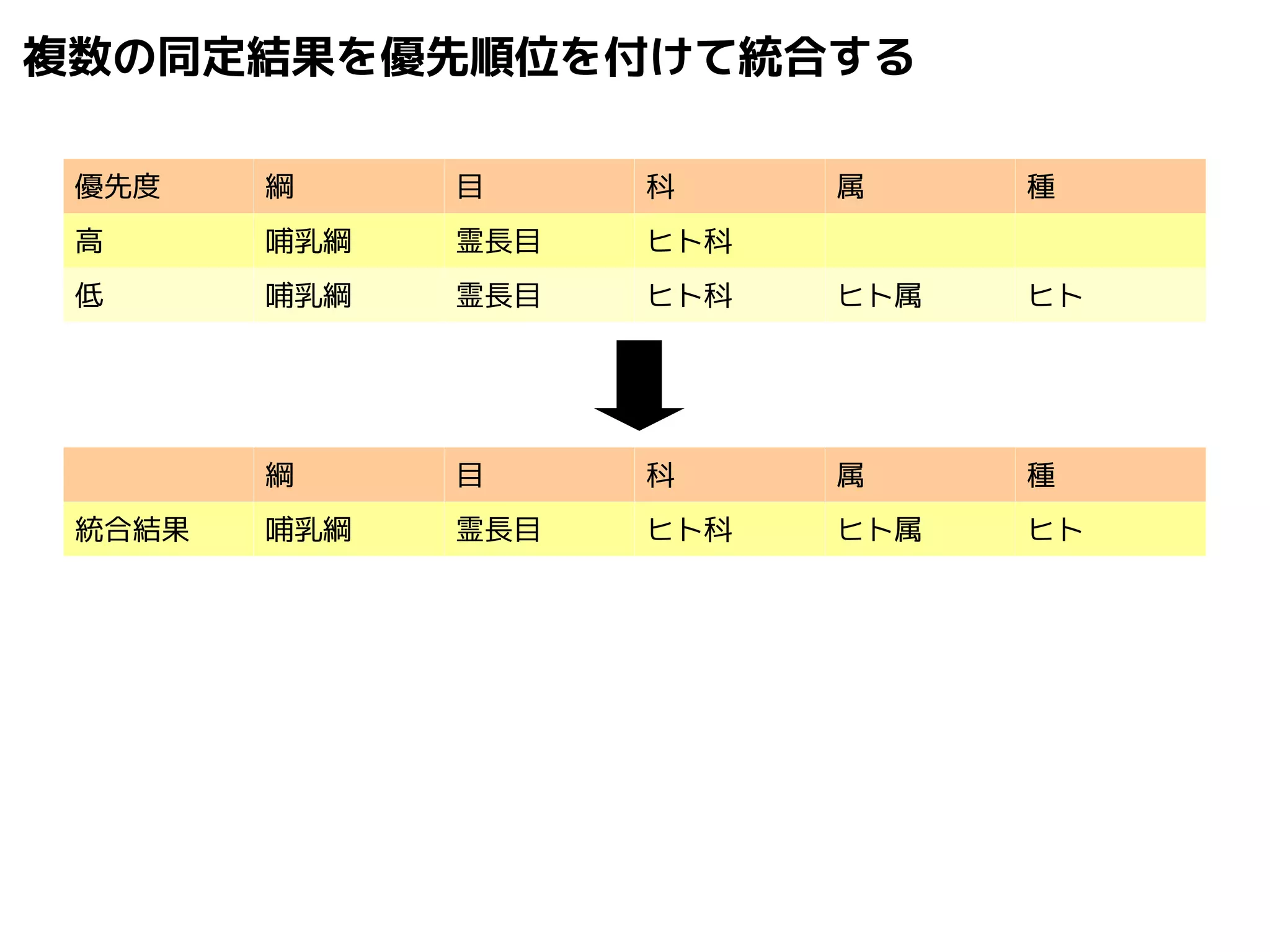































































![[DDBJing30] メタゲノム解析と微生物統合データベース](https://cdn.slidesharecdn.com/ss_thumbnails/30ddbjingmegap-141226013548-conversion-gate01-thumbnail.jpg?width=640&height=640&fit=bounds)













![[2019-09-02] AI・IoT活用情報とGoogle Colab植物画像注釈](https://cdn.slidesharecdn.com/ss_thumbnails/plantinformatics190902-190906044014-thumbnail.jpg?width=640&height=640&fit=bounds)













