Download free for 30 days
Sign in
Upload
Language (EN)
Support
Business
Mobile
Social Media
Marketing
Technology
Art & Photos
Career
Design
Education
Presentations & Public Speaking
Government & Nonprofit
Healthcare
Internet
Law
Leadership & Management
Automotive
Engineering
Software
Recruiting & HR
Retail
Sales
Services
Science
Small Business & Entrepreneurship
Food
Environment
Economy & Finance
Data & Analytics
Investor Relations
Sports
Spiritual
News & Politics
Travel
Self Improvement
Real Estate
Entertainment & Humor
Health & Medicine
Devices & Hardware
Lifestyle
Change Language
Language
English
Español
Português
Français
Deutsche
Cancel
Save
Submit search
EN
Uploaded by
Amelieff
27,559 views
Exome解析入門
2012年7月7日に開催した、アメリエフ株式会社・第14回勉強会「Exome解析入門」のスライドです。
Health & Medicine
◦
Read more
17
Save
Share
Embed
Embed presentation
1
/ 25
2
/ 25
Most read
3
/ 25
Most read
4
/ 25
Most read
5
/ 25
6
/ 25
7
/ 25
8
/ 25
9
/ 25
10
/ 25
11
/ 25
12
/ 25
13
/ 25
14
/ 25
15
/ 25
16
/ 25
17
/ 25
18
/ 25
19
/ 25
20
/ 25
21
/ 25
22
/ 25
23
/ 25
24
/ 25
25
/ 25
More Related Content
PDF
フリーソフトで始めるNGS解析_第41・42回勉強会資料
by
Amelieff
PDF
CBI学会2013チュートリアル NGSデータ解析入門 (解析編)配布資料
by
Genaris Omics, Inc.
PPTX
ゲノム育種を実装・利用するためのNGSデータ解析
by
Hiromi Kajiya-Kanegae
PDF
NGSを用いたジェノタイピングを様々な解析に用いるには?
by
Hiromi Kajiya-Kanegae
PDF
フリーソフトではじめるがん体細胞変異解析入門 第33回勉強会資料
by
Amelieff
PDF
[DDBJing33] ゲノムワイド多型を利用した遺伝解析の実際
by
DNA Data Bank of Japan center
PPTX
[DL輪読会]FOTS: Fast Oriented Text Spotting with a Unified Network
by
Deep Learning JP
PDF
フリーソフトではじめるNGS融合遺伝子解析入門
by
Amelieff
フリーソフトで始めるNGS解析_第41・42回勉強会資料
by
Amelieff
CBI学会2013チュートリアル NGSデータ解析入門 (解析編)配布資料
by
Genaris Omics, Inc.
ゲノム育種を実装・利用するためのNGSデータ解析
by
Hiromi Kajiya-Kanegae
NGSを用いたジェノタイピングを様々な解析に用いるには?
by
Hiromi Kajiya-Kanegae
フリーソフトではじめるがん体細胞変異解析入門 第33回勉強会資料
by
Amelieff
[DDBJing33] ゲノムワイド多型を利用した遺伝解析の実際
by
DNA Data Bank of Japan center
[DL輪読会]FOTS: Fast Oriented Text Spotting with a Unified Network
by
Deep Learning JP
フリーソフトではじめるNGS融合遺伝子解析入門
by
Amelieff
What's hot
PDF
NGS現場の会第2回_アメリエフ株式会社_がんExome解析
by
Amelieff
PPTX
第五回統計学勉強会@東大駒場
by
Daisuke Yoneoka
PDF
経験ベイズ木(IBIS 2017)
by
Masashi Sekino
PDF
Fiberの使いどころ
by
Tomoya Kawanishi
PDF
SNPデータ解析入門
by
Amelieff
PDF
機械学習のためのベイズ最適化入門
by
hoxo_m
PDF
混合整数ブラックボックス最適化に向けたCMA-ESの改良 / Optuna Meetup #2
by
RHamano
PDF
NGS現場の会第4回研究会 モーニング教育セッション 配布用資料 「Windows/Mac環境で始めるNGSデータ解析入門」
by
Genaris Omics, Inc.
PPTX
Rで因子分析 商用ソフトで実行できない因子分析のあれこれ
by
Hiroshi Shimizu
PPTX
Next Gen Sequencing (NGS) Technology Overview
by
Dominic Suciu
PPTX
[DL輪読会]Flow-based Deep Generative Models
by
Deep Learning JP
PDF
[DL輪読会]data2vec: A General Framework for Self-supervised Learning in Speech,...
by
Deep Learning JP
PDF
変分推論法(変分ベイズ法)(PRML第10章)
by
Takao Yamanaka
PPTX
未出現事象の出現確率
by
Hiroshi Nakagawa
PPTX
[DL輪読会]Learning convolutional neural networks for graphs
by
Deep Learning JP
PDF
EMアルゴリズム
by
Sotetsu KOYAMADA(小山田創哲)
PDF
文脈自由文法の話
by
kogecoo
PPTX
PyTorchLightning ベース Hydra+MLFlow+Optuna による機械学習開発環境の構築
by
Kosuke Shinoda
PDF
TVM の紹介
by
Masahiro Masuda
PPTX
高速なガンマ分布の最尤推定法について
by
hoxo_m
NGS現場の会第2回_アメリエフ株式会社_がんExome解析
by
Amelieff
第五回統計学勉強会@東大駒場
by
Daisuke Yoneoka
経験ベイズ木(IBIS 2017)
by
Masashi Sekino
Fiberの使いどころ
by
Tomoya Kawanishi
SNPデータ解析入門
by
Amelieff
機械学習のためのベイズ最適化入門
by
hoxo_m
混合整数ブラックボックス最適化に向けたCMA-ESの改良 / Optuna Meetup #2
by
RHamano
NGS現場の会第4回研究会 モーニング教育セッション 配布用資料 「Windows/Mac環境で始めるNGSデータ解析入門」
by
Genaris Omics, Inc.
Rで因子分析 商用ソフトで実行できない因子分析のあれこれ
by
Hiroshi Shimizu
Next Gen Sequencing (NGS) Technology Overview
by
Dominic Suciu
[DL輪読会]Flow-based Deep Generative Models
by
Deep Learning JP
[DL輪読会]data2vec: A General Framework for Self-supervised Learning in Speech,...
by
Deep Learning JP
変分推論法(変分ベイズ法)(PRML第10章)
by
Takao Yamanaka
未出現事象の出現確率
by
Hiroshi Nakagawa
[DL輪読会]Learning convolutional neural networks for graphs
by
Deep Learning JP
EMアルゴリズム
by
Sotetsu KOYAMADA(小山田創哲)
文脈自由文法の話
by
kogecoo
PyTorchLightning ベース Hydra+MLFlow+Optuna による機械学習開発環境の構築
by
Kosuke Shinoda
TVM の紹介
by
Masahiro Masuda
高速なガンマ分布の最尤推定法について
by
hoxo_m
Similar to Exome解析入門
PDF
NGS現場の会第2回_アメリエフ株式会社_Qcleaner
by
Amelieff
PDF
フリーソフトではじめるChIP-seq解析_第40回勉強会資料
by
Amelieff
PDF
NGS現場の会第2回_アメリエフ株式会社_RNAseq解析
by
Amelieff
PDF
20160324自由集会講演
by
astanabe
PDF
RNASkim
by
弘毅 露崎
PDF
次世代シーケンス解析サーバーReseq解析マニュアル
by
Amelieff
PDF
メタバーコーディングのフレームワークとアルゴリズム
by
astanabe
PDF
フリーソフトではじめるメチル化データ解析入門 SeqCap Epiデータ対応_第40回勉強会資料
by
Amelieff
PPTX
BGI Webinar Aug 28, 2014 "Genome wide methylation analysis and analytics"
by
kazuoishii20
PPTX
2014年度 第5回バイオインフォマティクス実習
by
Jun Nakabayashi
PDF
第9回 配信講義 計算科学技術特論B(2022)
by
RCCSRENKEI
PDF
新規医療開発に関わる統計学 (バイオインフォマティクス)
by
Hidemasa Bono
PDF
生命化学情報学2
by
Mas Kot
PPTX
2019年度 第2回バイオインフォマティクス実習
by
Jun Nakabayashi
PPTX
2015年度 第2回バイオインフォマティクス実習
by
Jun Nakabayashi
PDF
miRNAデータ解析入門_第23回勉強会資料
by
Amelieff
PDF
ISMB/ECCB2015読み会イントロ+Misassembly detection using paired-end sequence reads an...
by
Kengo Sato
PDF
環境試料からの核酸精製キット ミニカタログ(ZYR : ZYMO RESEARCH/#5764) | フナコシ株式会社
by
fu7koshi
PPTX
2016年度 第4回バイオインフォマティクス実習
by
Jun Nakabayashi
PDF
drbonodojo3-6 データ統合解析
by
Yoshiaki Yasumizu
NGS現場の会第2回_アメリエフ株式会社_Qcleaner
by
Amelieff
フリーソフトではじめるChIP-seq解析_第40回勉強会資料
by
Amelieff
NGS現場の会第2回_アメリエフ株式会社_RNAseq解析
by
Amelieff
20160324自由集会講演
by
astanabe
RNASkim
by
弘毅 露崎
次世代シーケンス解析サーバーReseq解析マニュアル
by
Amelieff
メタバーコーディングのフレームワークとアルゴリズム
by
astanabe
フリーソフトではじめるメチル化データ解析入門 SeqCap Epiデータ対応_第40回勉強会資料
by
Amelieff
BGI Webinar Aug 28, 2014 "Genome wide methylation analysis and analytics"
by
kazuoishii20
2014年度 第5回バイオインフォマティクス実習
by
Jun Nakabayashi
第9回 配信講義 計算科学技術特論B(2022)
by
RCCSRENKEI
新規医療開発に関わる統計学 (バイオインフォマティクス)
by
Hidemasa Bono
生命化学情報学2
by
Mas Kot
2019年度 第2回バイオインフォマティクス実習
by
Jun Nakabayashi
2015年度 第2回バイオインフォマティクス実習
by
Jun Nakabayashi
miRNAデータ解析入門_第23回勉強会資料
by
Amelieff
ISMB/ECCB2015読み会イントロ+Misassembly detection using paired-end sequence reads an...
by
Kengo Sato
環境試料からの核酸精製キット ミニカタログ(ZYR : ZYMO RESEARCH/#5764) | フナコシ株式会社
by
fu7koshi
2016年度 第4回バイオインフォマティクス実習
by
Jun Nakabayashi
drbonodojo3-6 データ統合解析
by
Yoshiaki Yasumizu
Exome解析入門
1.
Exomeデータ解析入門
2012年7月7日 アメリエフ株式会社 Copyright © Amelieff Co. Ltd. All Rights Reserved.
2.
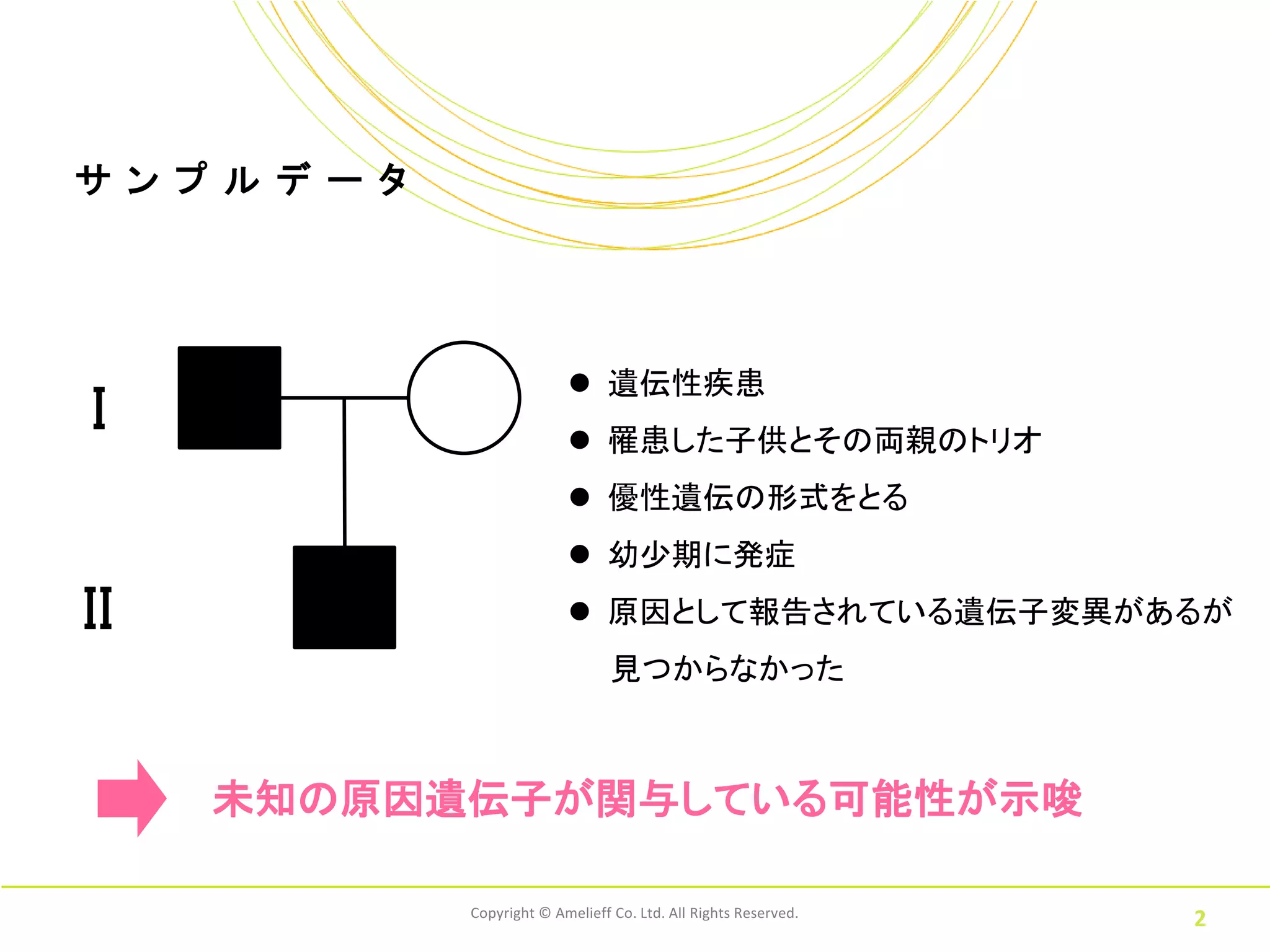
サ ン プ
ル デ ー タ 遺伝性疾患 I 罹患した子供とその両親のトリオ 優性遺伝の形式をとる 幼少期に発症 II 原因として報告されている遺伝子変異があるが 見つからなかった 未知の原因遺伝子が関与している可能性が示唆 Copyright © Amelieff Co. Ltd. All Rights Reserved. 2
3.
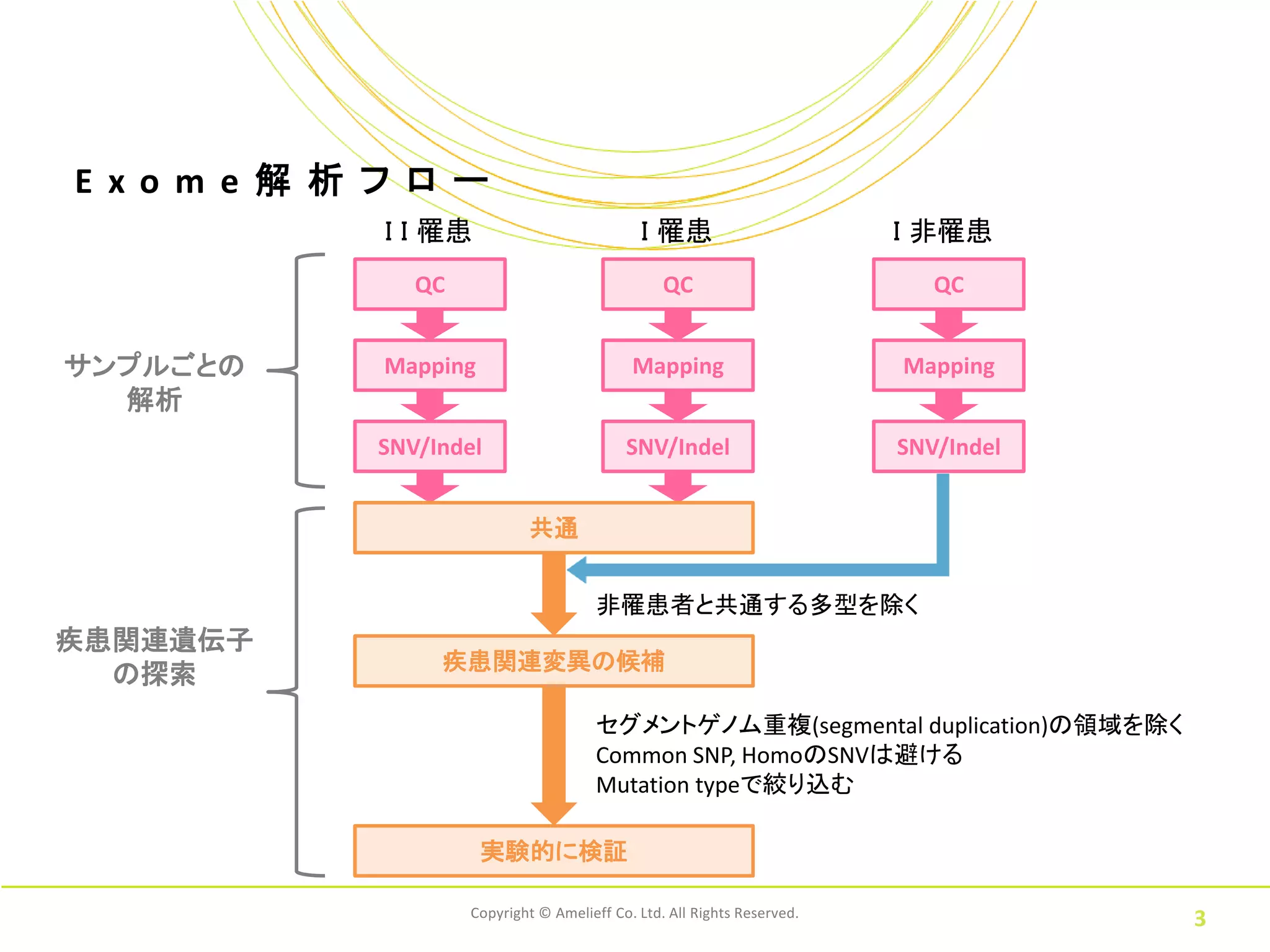
E xo m
e 解 析 フ ロ ー I I 罹患 I 罹患 I 非罹患 QC QC QC サンプルごとの Mapping Mapping Mapping 解析 SNV/Indel SNV/Indel SNV/Indel 共通 非罹患者と共通する多型を除く 疾患関連遺伝子 疾患関連変異の候補 の探索 セグメントゲノム重複(segmental duplication)の領域を除く Common SNP, HomoのSNVは避ける Mutation typeで絞り込む 実験的に検証 Copyright © Amelieff Co. Ltd. All Rights Reserved. 3
4.
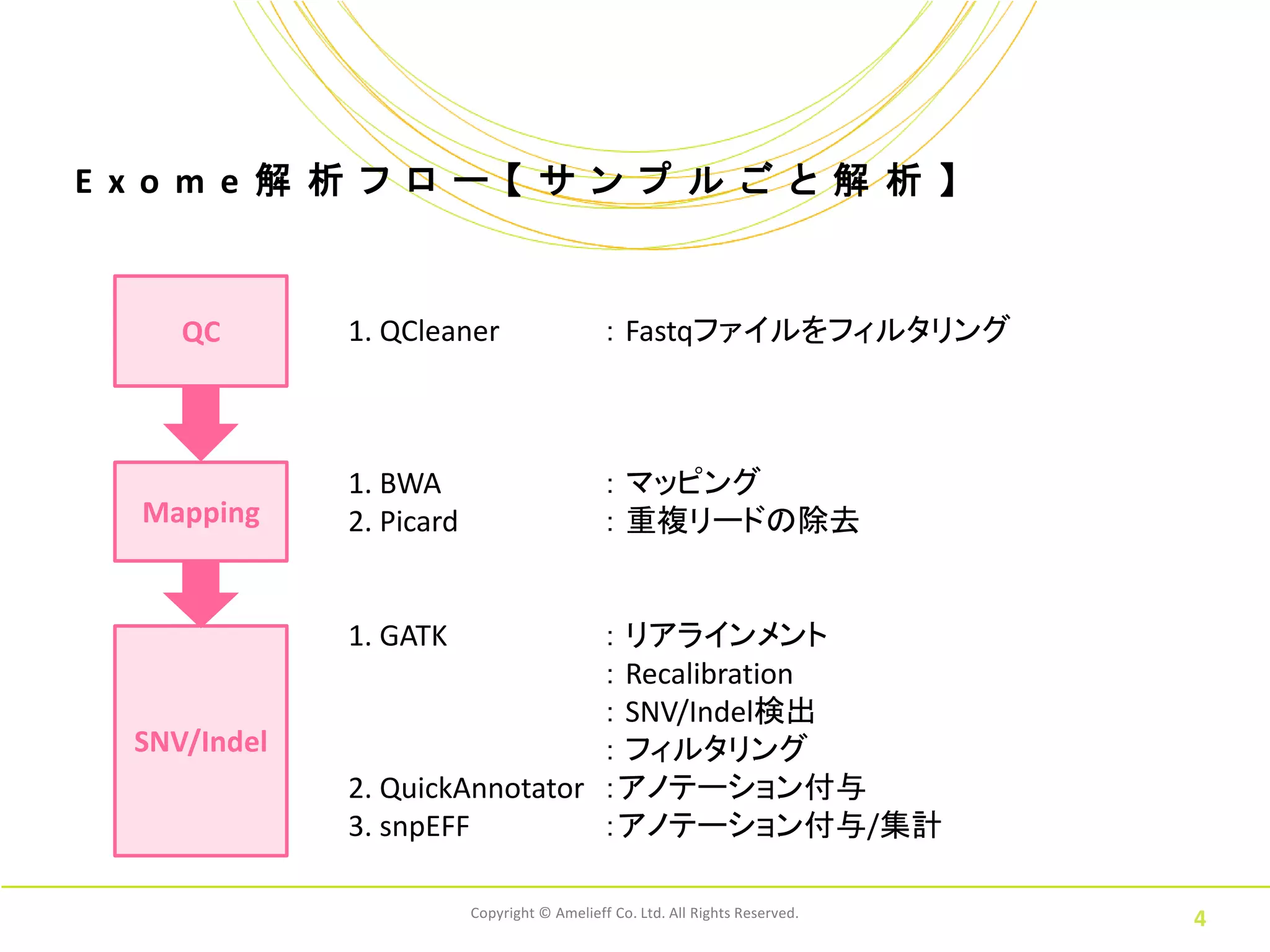
E xo m
e 解 析 フ ロ ー 【 サ ン プ ル ご と 解 析 】 QC 1. QCleaner : Fastqファイルをフィルタリング 1. BWA : マッピング Mapping 2. Picard : 重複リードの除去 1. GATK : リアラインメント : Recalibration : SNV/Indel検出 SNV/Indel : フィルタリング 2. QuickAnnotator :アノテーション付与 3. snpEFF :アノテーション付与/集計 Copyright © Amelieff Co. Ltd. All Rights Reserved. 4
5.
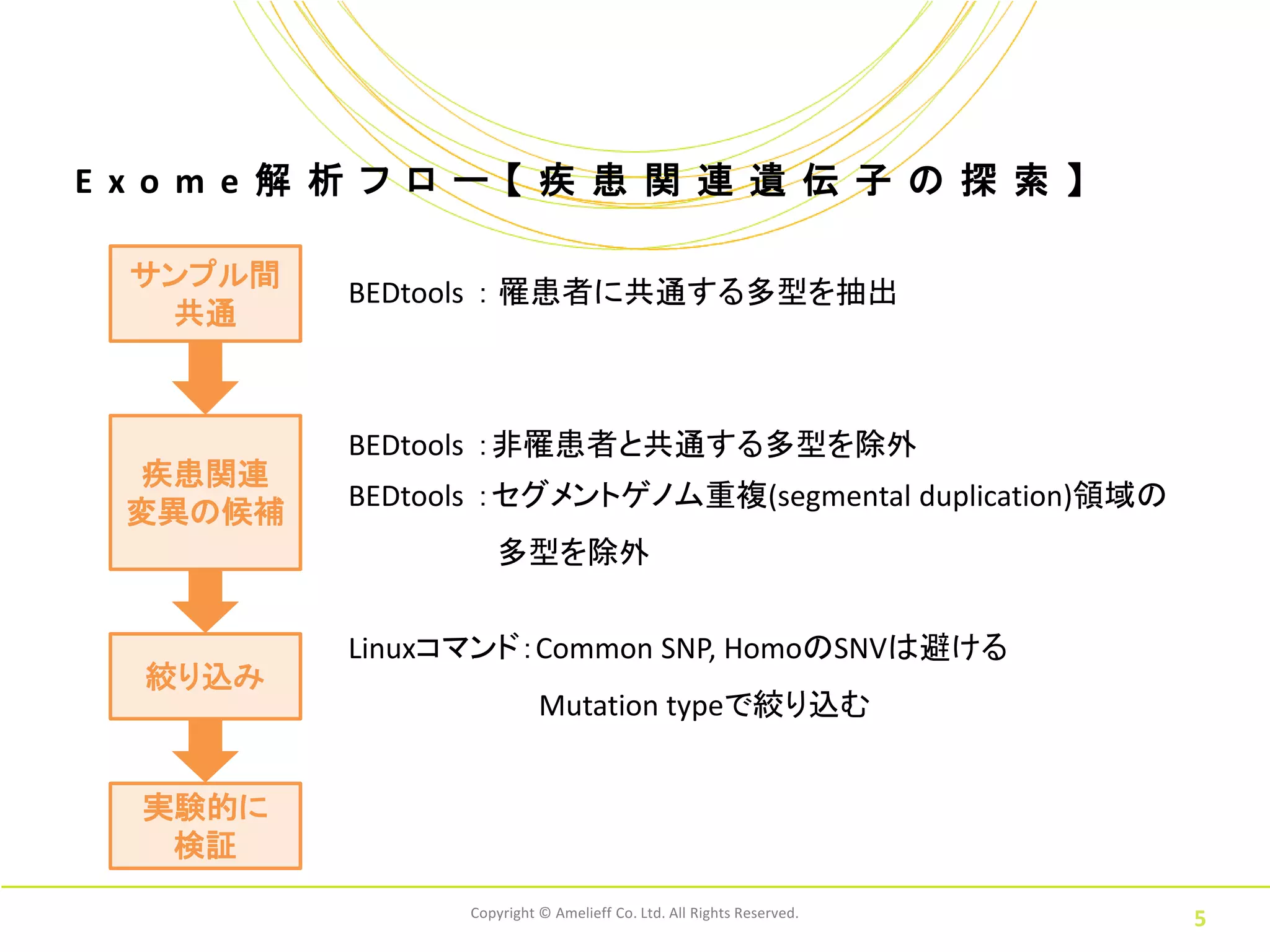
E xo m
e 解 析 フ ロ ー 【 疾 患 関 連 遺 伝 子 の 探 索 】 サンプル間 BEDtools : 罹患者に共通する多型を抽出 共通 BEDtools :非罹患者と共通する多型を除外 疾患関連 BEDtools :セグメントゲノム重複(segmental duplication)領域の 変異の候補 多型を除外 Linuxコマンド:Common SNP, HomoのSNVは避ける 絞り込み Mutation typeで絞り込む 実験的に 検証 Copyright © Amelieff Co. Ltd. All Rights Reserved. 5
6.
サ ン プ
ル ご と の 解 析 【コマンド】 1. QCleaner : Fastqファイルをフィルタリング ※自社開発のプログラムです。 QC $ perl qcleaner.pl --i1 $FILE1.fastq --i2 $FILE2.fastq --o1 $FILE1.clean.fastq --o2 $FILE2.clean.fastq --qp 20,80 --n 10 -- trim 20 --length 20 --log $FILE.qc.log FASTQ形式にマッチするかチェック データクオリティチェック(FastQC) Mapping Illumina CASAVA filter [Y] を除去 クオリティ20未満が80%以上のリードを除去 クオリティ20未満の末端をトリム 未知の塩基(N)が多いリード除去 SNV/Indel 配列長が短いリード除去 片側のみのリードを除外 データクオリティチェック(FastQC) Copyright © Amelieff Co. Ltd. All Rights Reserved. 6
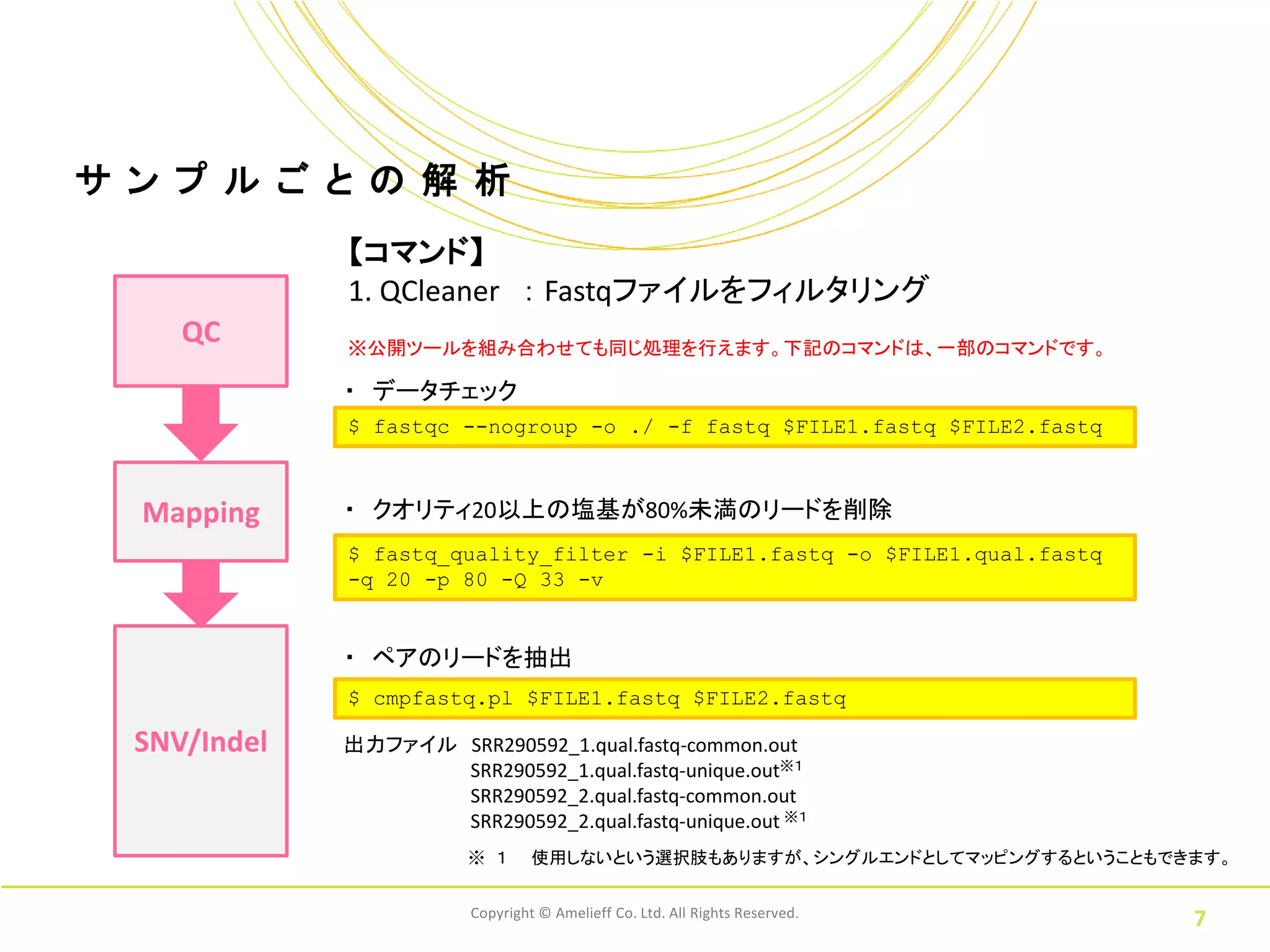
7.
サ ン プ
ル ご と の 解 析 【コマンド】 1. QCleaner : Fastqファイルをフィルタリング QC ※公開ツールを組み合わせても同じ処理を行えます。下記のコマンドは、一部のコマンドです。 ・ データチェック $ fastqc --nogroup -o ./ -f fastq $FILE1.fastq $FILE2.fastq Mapping ・ クオリティ20以上の塩基が80%未満のリードを削除 $ fastq_quality_filter -i $FILE1.fastq -o $FILE1.qual.fastq -q 20 -p 80 -Q 33 -v ・ ペアのリードを抽出 $ cmpfastq.pl $FILE1.fastq $FILE2.fastq SNV/Indel 出力ファイル SRR290592_1.qual.fastq-common.out SRR290592_1.qual.fastq-unique.out※1 SRR290592_2.qual.fastq-common.out SRR290592_2.qual.fastq-unique.out ※1 ※ 1 使用しないという選択肢もありますが、シングルエンドとしてマッピングするということもできます。 Copyright © Amelieff Co. Ltd. All Rights Reserved. 7
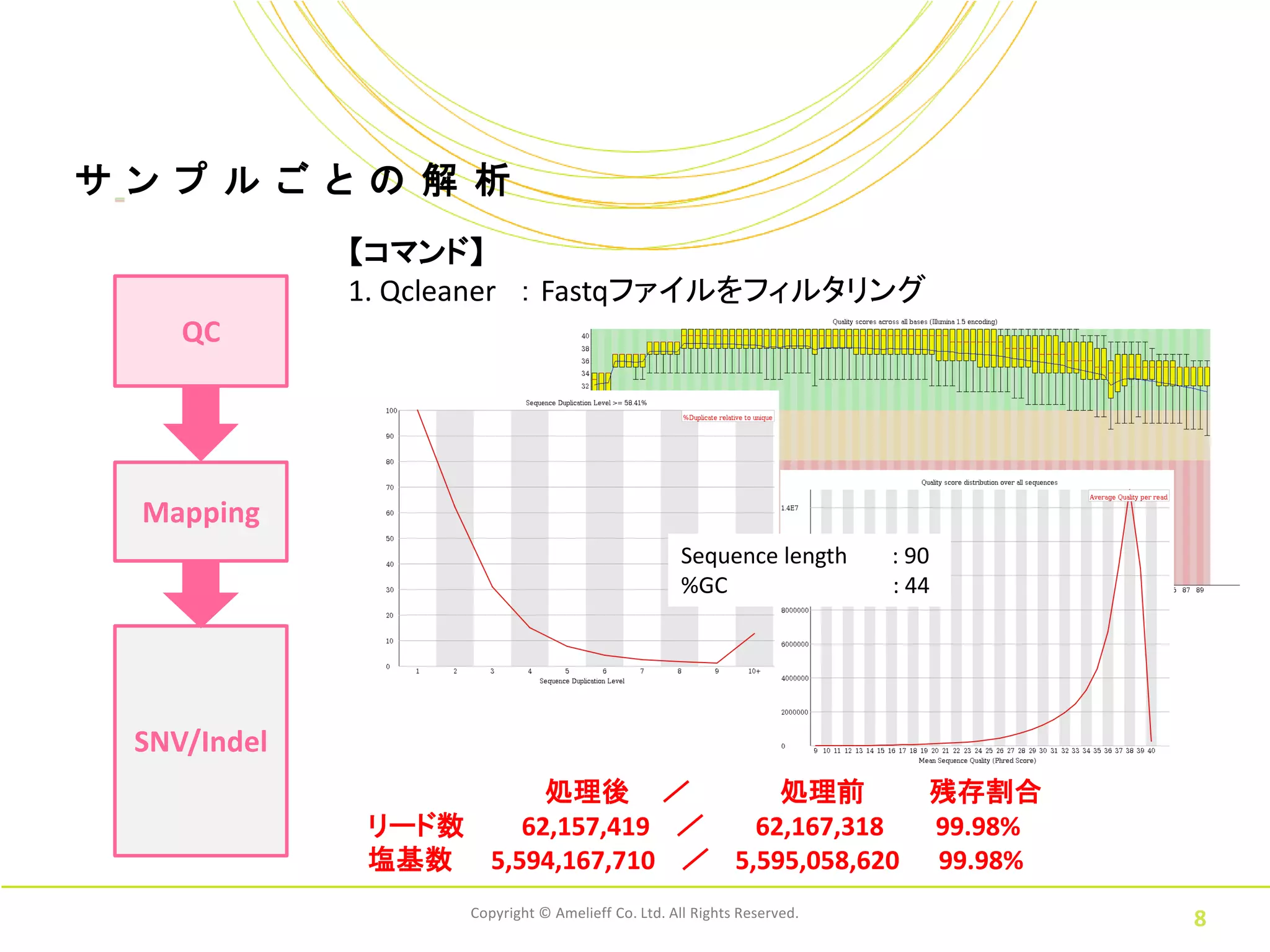
8.
サ ン プ
ル ご と の 解 析 【コマンド】 1. Qcleaner : Fastqファイルをフィルタリング QC Mapping Sequence length : 90 %GC : 44 SNV/Indel 処理後 / 処理前 残存割合 リード数 62,157,419 / 62,167,318 99.98% 塩基数 5,594,167,710 / 5,595,058,620 99.98% Copyright © Amelieff Co. Ltd. All Rights Reserved. 8
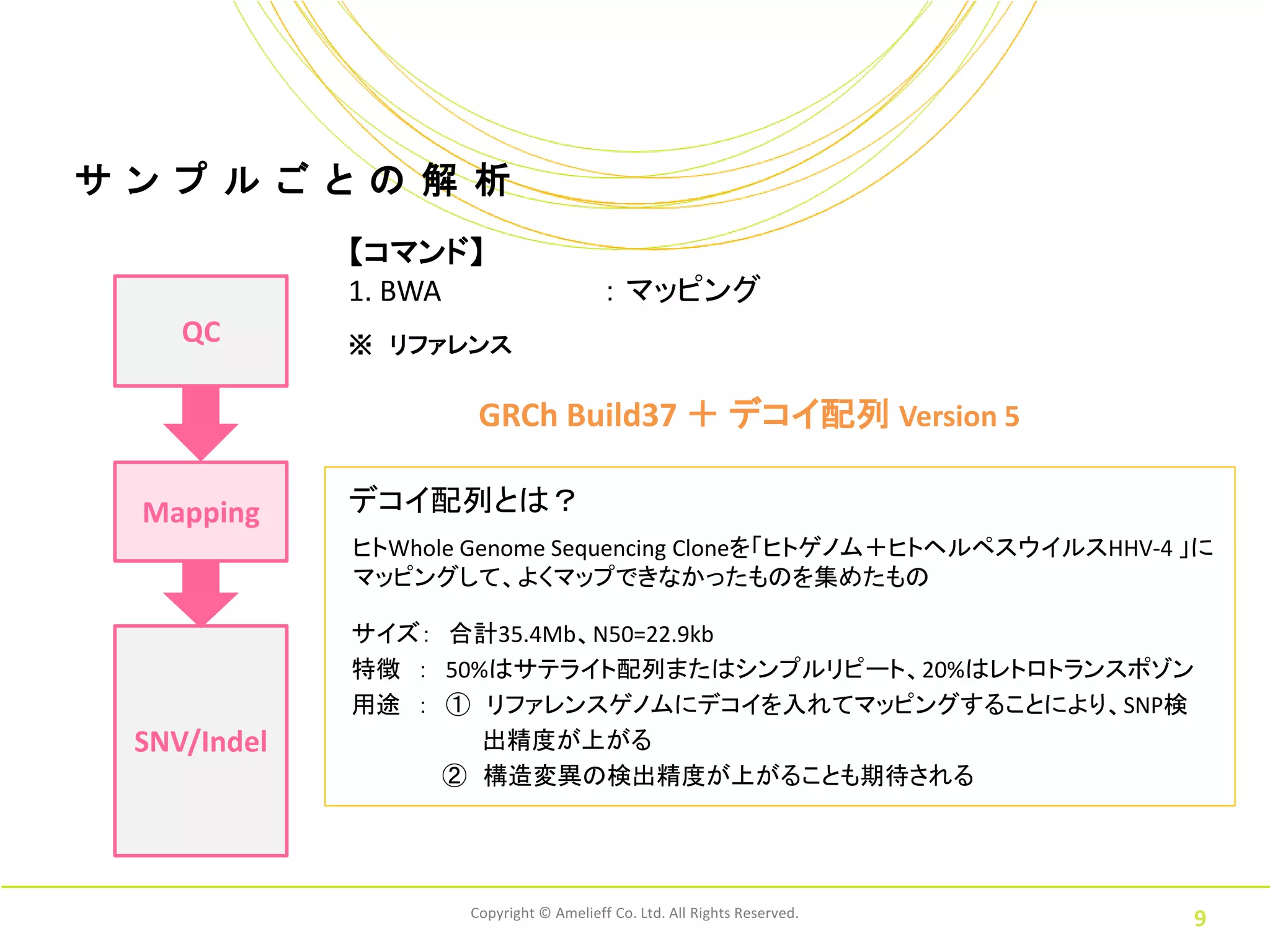
9.
サ ン プ
ル ご と の 解 析 【コマンド】 1. BWA : マッピング QC ※ リファレンス GRCh Build37 + デコイ配列 Version 5 Mapping デコイ配列とは? ヒトWhole Genome Sequencing Cloneを「ヒトゲノム+ヒトヘルペスウイルスHHV-4 」に マッピングして、よくマップできなかったものを集めたもの サイズ: 合計35.4Mb、N50=22.9kb 特徴 : 50%はサテライト配列またはシンプルリピート、20%はレトロトランスポゾン 用途 : ① リファレンスゲノムにデコイを入れてマッピングすることにより、SNP検 SNV/Indel 出精度が上がる ② 構造変異の検出精度が上がることも期待される Copyright © Amelieff Co. Ltd. All Rights Reserved. 9
10.
サ ン プ
ル ご と の 解 析 【コマンド】 1. BWA : マッピング QC ・ マッピング/SAMをBAMに変換/ソート $ bwa aln -t 6 genome_hs37d5cs.fa $FILE1.clean.fastq -f $FILE1.sai $ bwa sampe -r "@RG¥tID:$FILE¥tSM:$FILE¥tPL:Illumina" -n 3 Mapping -N 10 -a 500 genome_hs37d5cs.fa $FILE1.sai $FILE2.sai $FILE1.clean.fastq $FILE2.clean.fastq | samtools view -Sb - | samtools sort - $FILE.sorted ※オプション -a INT maximum insert size [500] -n INT maximum hits to output for paired reads [3] -N INT maximum hits to output for discordant pairs [10] SNV/Indel ※RG(read groups) platform (PL) および sample (SM)が必要 PLの例:454, LS454, Illumina, Solid, ABI_Solid, CG (all case-insensitive) 解析ツールGATKに入力するBAMファイルに、RGタグの記述がないとエラーが出る Copyright © Amelieff Co. Ltd. All Rights Reserved. 10
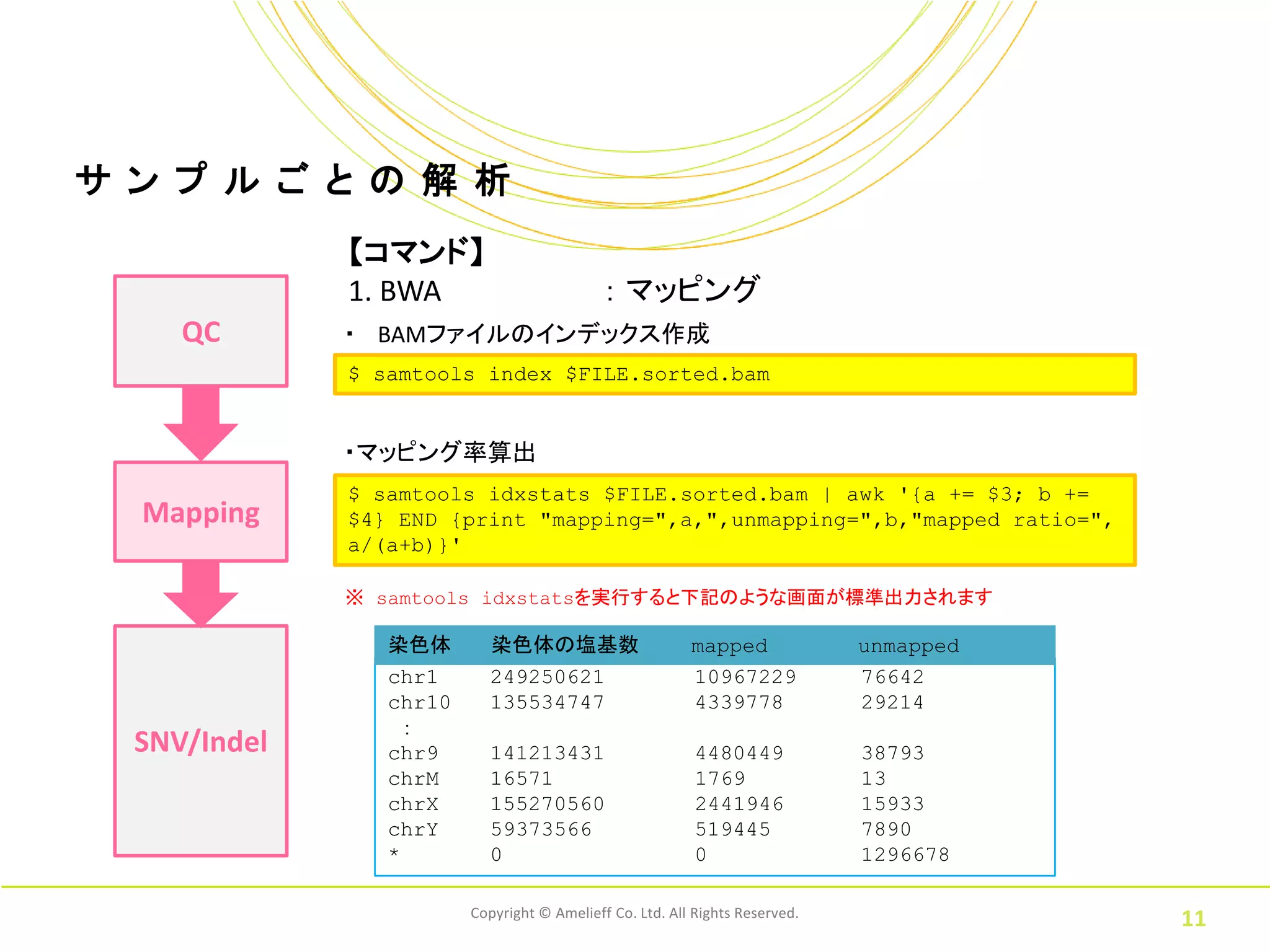
11.
サ ン プ
ル ご と の 解 析 【コマンド】 1. BWA : マッピング QC ・ BAMファイルのインデックス作成 $ samtools index $FILE.sorted.bam ・マッピング率算出 $ samtools idxstats $FILE.sorted.bam | awk '{a += $3; b += Mapping $4} END {print "mapping=",a,",unmapping=",b,"mapped ratio=", a/(a+b)}' ※ samtools idxstatsを実行すると下記のような画面が標準出力されます 染色体 染色体の塩基数 mapped unmapped chr1 249250621 10967229 76642 chr10 135534747 4339778 29214 : SNV/Indel chr9 141213431 4480449 38793 chrM 16571 1769 13 chrX 155270560 2441946 15933 chrY 59373566 519445 7890 * 0 0 1296678 Copyright © Amelieff Co. Ltd. All Rights Reserved. 11
12.
サ ン プ
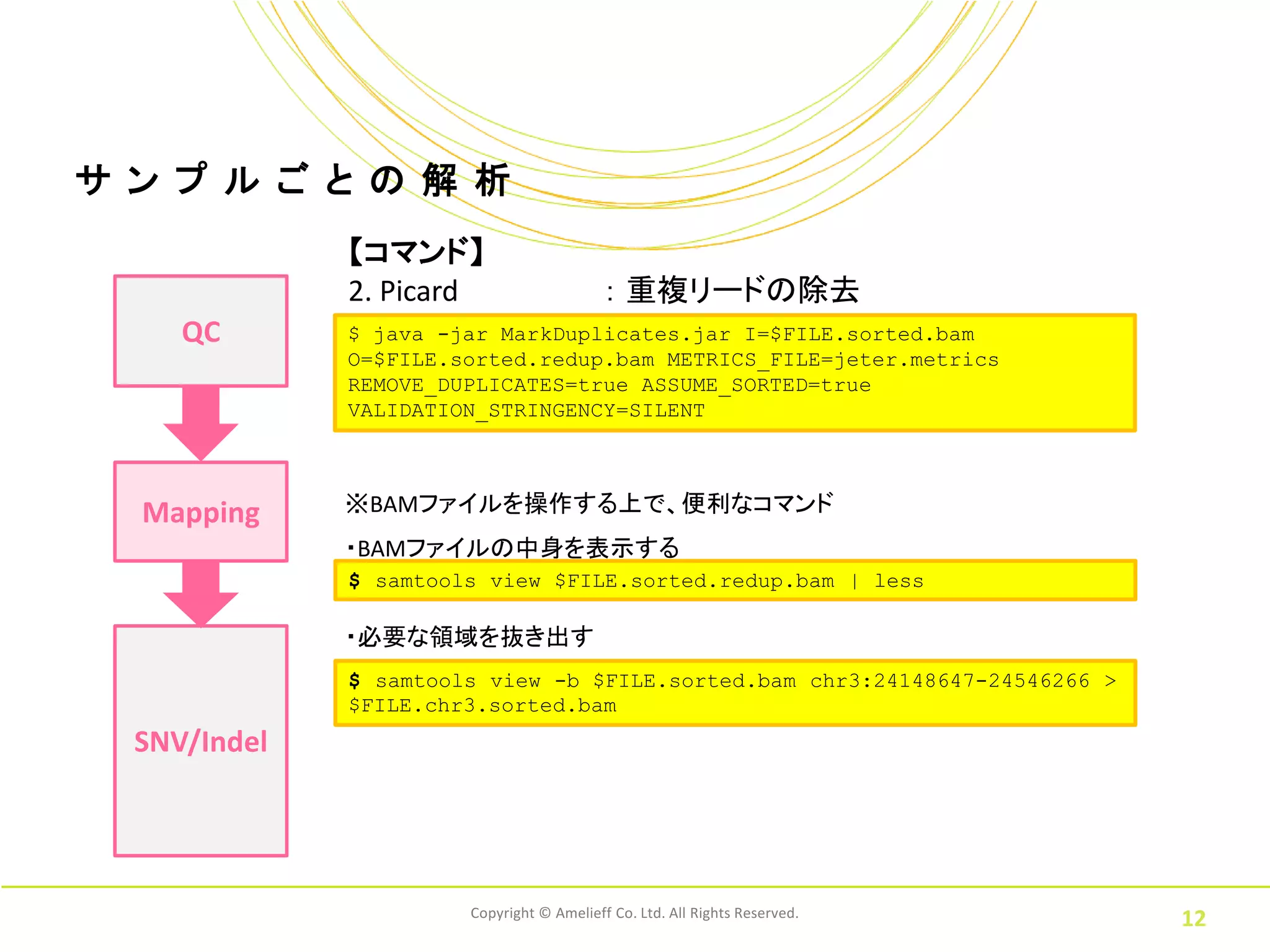
ル ご と の 解 析 【コマンド】 2. Picard : 重複リードの除去 QC $ java -jar MarkDuplicates.jar I=$FILE.sorted.bam O=$FILE.sorted.redup.bam METRICS_FILE=jeter.metrics REMOVE_DUPLICATES=true ASSUME_SORTED=true VALIDATION_STRINGENCY=SILENT Mapping ※BAMファイルを操作する上で、便利なコマンド ・BAMファイルの中身を表示する $ samtools view $FILE.sorted.redup.bam | less ・必要な領域を抜き出す $ samtools view -b $FILE.sorted.bam chr3:24148647-24546266 > $FILE.chr3.sorted.bam SNV/Indel Copyright © Amelieff Co. Ltd. All Rights Reserved. 12
13.
サ ン プ
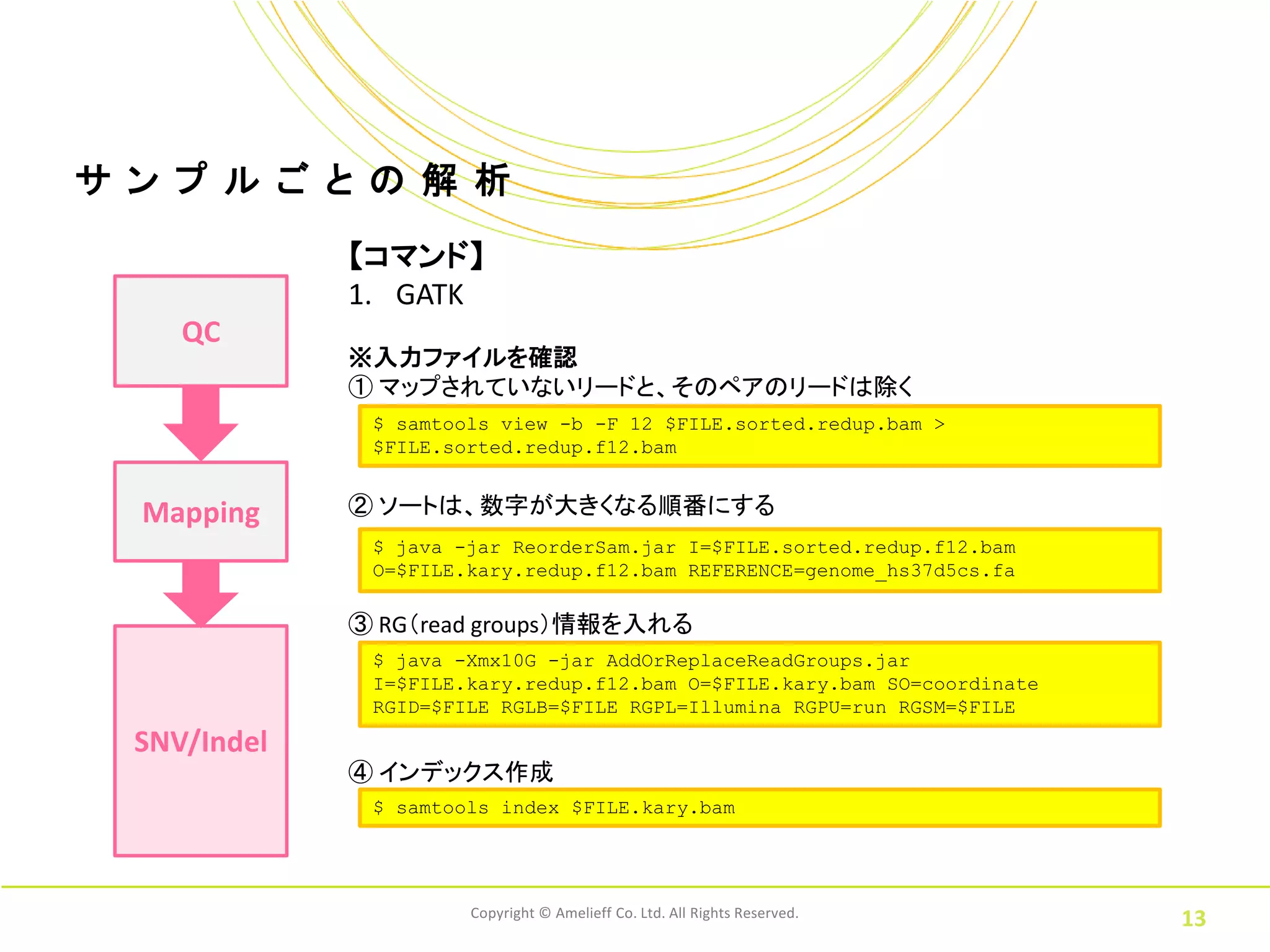
ル ご と の 解 析 【コマンド】 1. GATK QC ※入力ファイルを確認 ① マップされていないリードと、そのペアのリードは除く $ samtools view -b -F 12 $FILE.sorted.redup.bam > $FILE.sorted.redup.f12.bam Mapping ② ソートは、数字が大きくなる順番にする $ java -jar ReorderSam.jar I=$FILE.sorted.redup.f12.bam O=$FILE.kary.redup.f12.bam REFERENCE=genome_hs37d5cs.fa ③ RG(read groups)情報を入れる $ java -Xmx10G -jar AddOrReplaceReadGroups.jar I=$FILE.kary.redup.f12.bam O=$FILE.kary.bam SO=coordinate RGID=$FILE RGLB=$FILE RGPL=Illumina RGPU=run RGSM=$FILE SNV/Indel ④ インデックス作成 $ samtools index $FILE.kary.bam Copyright © Amelieff Co. Ltd. All Rights Reserved. 13
14.
サ ン プ
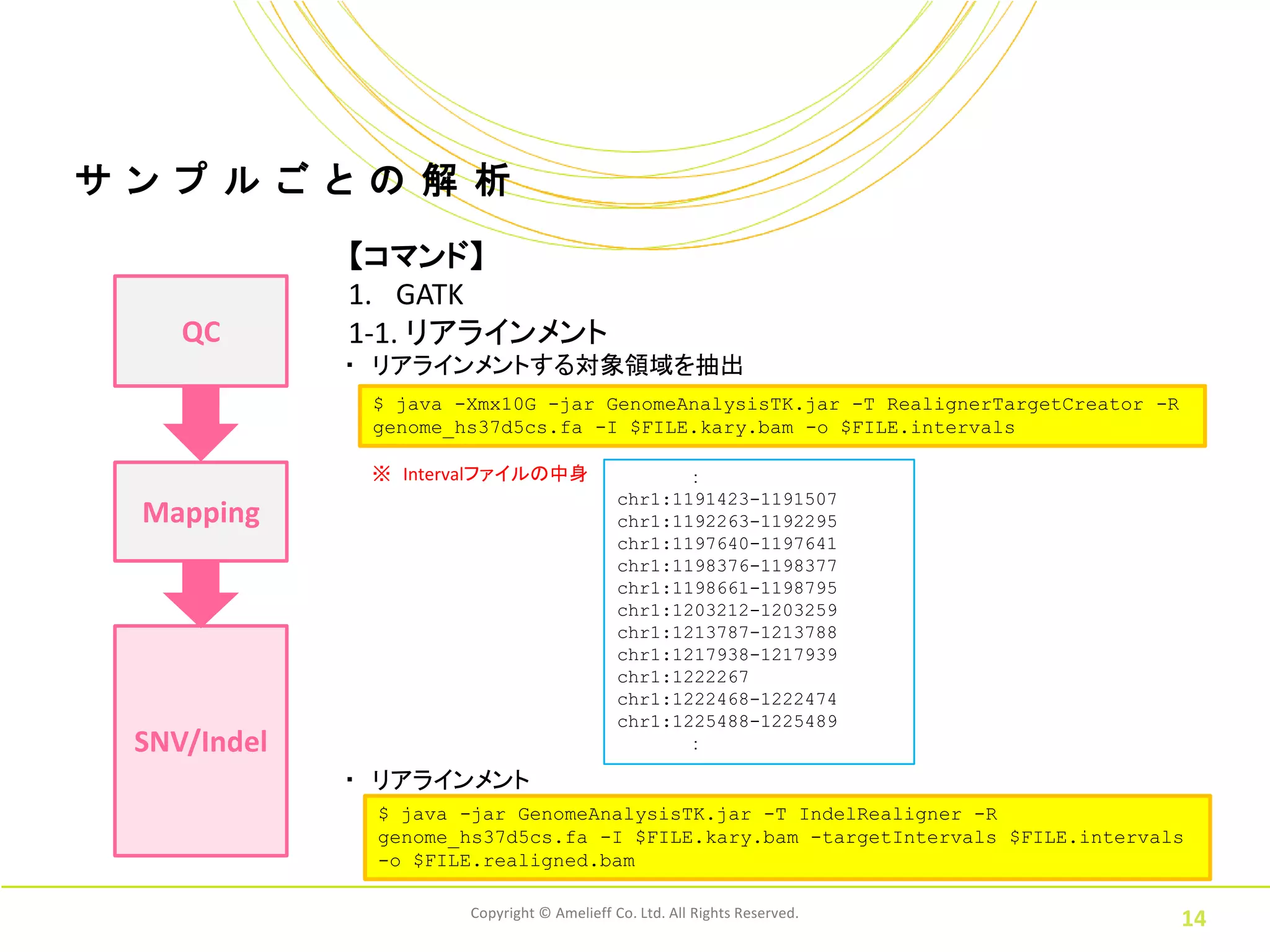
ル ご と の 解 析 【コマンド】 1. GATK QC 1-1. リアラインメント ・ リアラインメントする対象領域を抽出 $ java -Xmx10G -jar GenomeAnalysisTK.jar -T RealignerTargetCreator -R genome_hs37d5cs.fa -I $FILE.kary.bam -o $FILE.intervals ※ Intervalファイルの中身 : chr1:1191423-1191507 Mapping chr1:1192263-1192295 chr1:1197640-1197641 chr1:1198376-1198377 chr1:1198661-1198795 chr1:1203212-1203259 chr1:1213787-1213788 chr1:1217938-1217939 chr1:1222267 chr1:1222468-1222474 chr1:1225488-1225489 SNV/Indel : ・ リアラインメント $ java -jar GenomeAnalysisTK.jar -T IndelRealigner -R genome_hs37d5cs.fa -I $FILE.kary.bam -targetIntervals $FILE.intervals -o $FILE.realigned.bam Copyright © Amelieff Co. Ltd. All Rights Reserved. 14
15.
サ ン プ
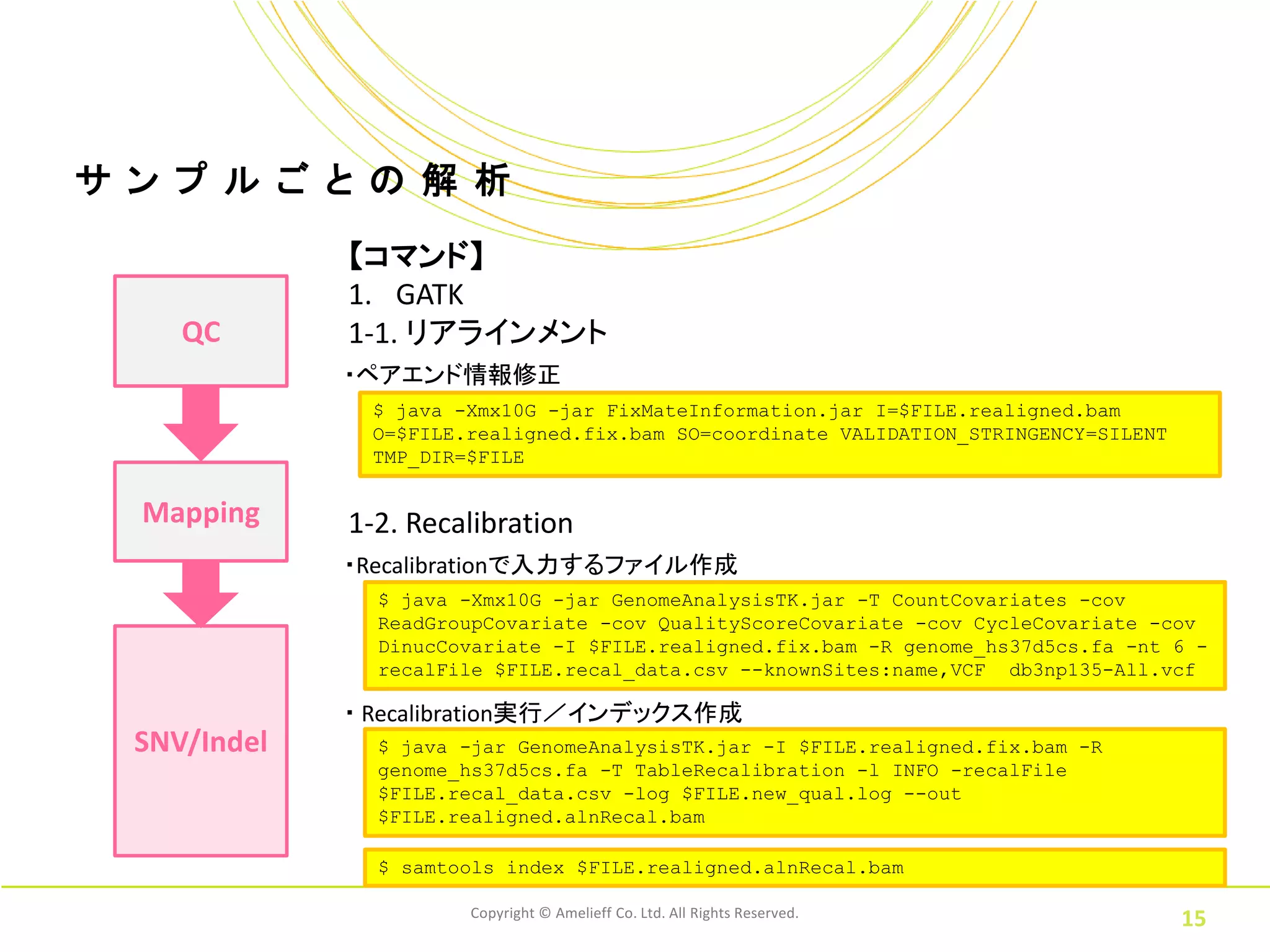
ル ご と の 解 析 【コマンド】 1. GATK QC 1-1. リアラインメント ・ペアエンド情報修正 $ java -Xmx10G -jar FixMateInformation.jar I=$FILE.realigned.bam O=$FILE.realigned.fix.bam SO=coordinate VALIDATION_STRINGENCY=SILENT TMP_DIR=$FILE Mapping 1-2. Recalibration ・Recalibrationで入力するファイル作成 $ java -Xmx10G -jar GenomeAnalysisTK.jar -T CountCovariates -cov ReadGroupCovariate -cov QualityScoreCovariate -cov CycleCovariate -cov DinucCovariate -I $FILE.realigned.fix.bam -R genome_hs37d5cs.fa -nt 6 - recalFile $FILE.recal_data.csv --knownSites:name,VCF db3np135-All.vcf ・ Recalibration実行/インデックス作成 SNV/Indel $ java -jar GenomeAnalysisTK.jar -I $FILE.realigned.fix.bam -R genome_hs37d5cs.fa -T TableRecalibration -l INFO -recalFile $FILE.recal_data.csv -log $FILE.new_qual.log --out $FILE.realigned.alnRecal.bam $ samtools index $FILE.realigned.alnRecal.bam Copyright © Amelieff Co. Ltd. All Rights Reserved. 15
16.
サ ン プ
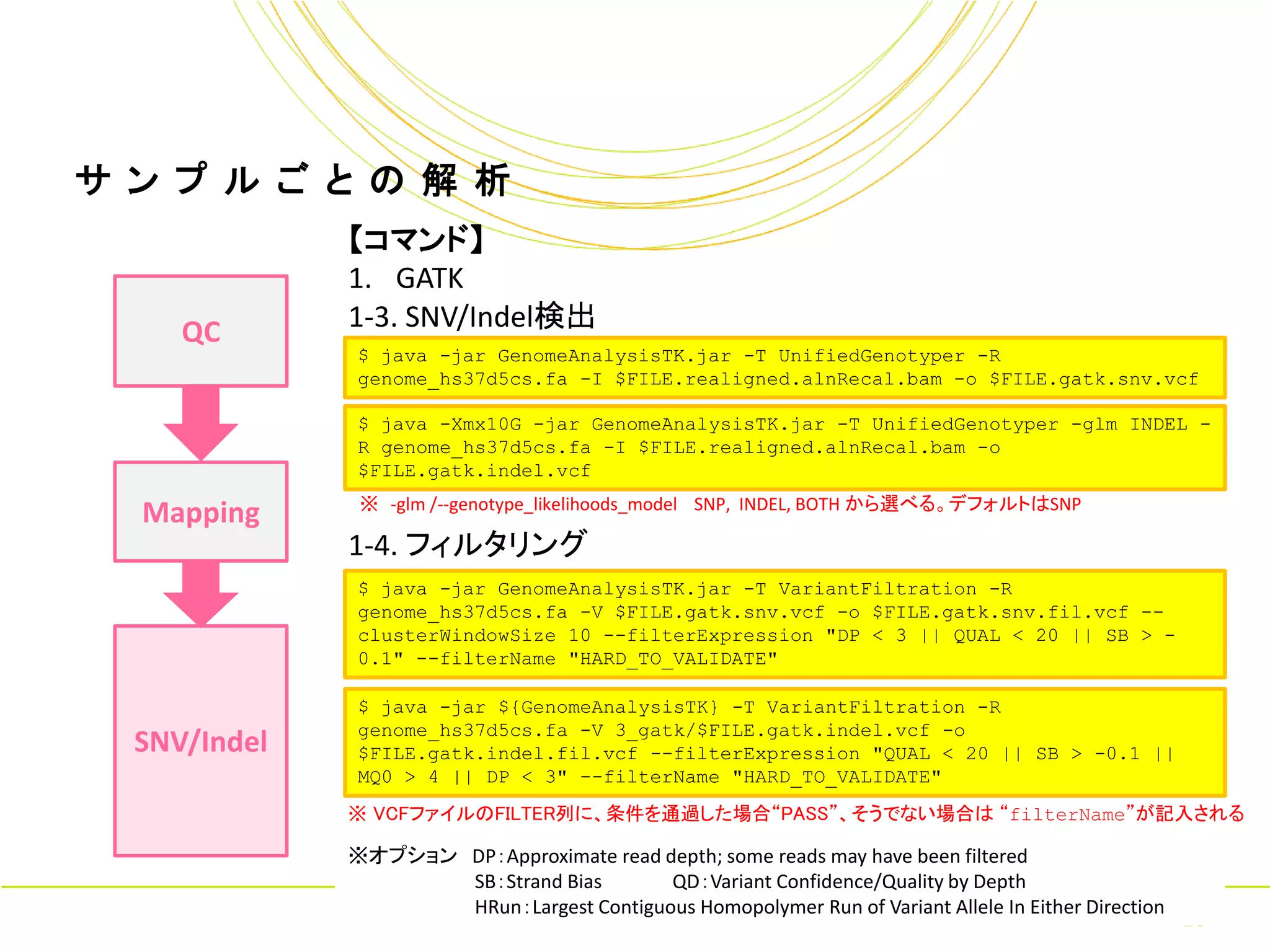
ル ご と の 解 析 【コマンド】 1. GATK QC 1-3. SNV/Indel検出 $ java -jar GenomeAnalysisTK.jar -T UnifiedGenotyper -R genome_hs37d5cs.fa -I $FILE.realigned.alnRecal.bam -o $FILE.gatk.snv.vcf $ java -Xmx10G -jar GenomeAnalysisTK.jar -T UnifiedGenotyper -glm INDEL - R genome_hs37d5cs.fa -I $FILE.realigned.alnRecal.bam -o $FILE.gatk.indel.vcf ※ -glm /--genotype_likelihoods_model SNP, INDEL, BOTH から選べる。デフォルトはSNP Mapping 1-4. フィルタリング $ java -jar GenomeAnalysisTK.jar -T VariantFiltration -R genome_hs37d5cs.fa -V $FILE.gatk.snv.vcf -o $FILE.gatk.snv.fil.vcf -- clusterWindowSize 10 --filterExpression "DP < 3 || QUAL < 20 || SB > - 0.1" --filterName "HARD_TO_VALIDATE" $ java -jar ${GenomeAnalysisTK} -T VariantFiltration -R genome_hs37d5cs.fa -V 3_gatk/$FILE.gatk.indel.vcf -o SNV/Indel $FILE.gatk.indel.fil.vcf --filterExpression "QUAL < 20 || SB > -0.1 || MQ0 > 4 || DP < 3" --filterName "HARD_TO_VALIDATE" ※ VCFファイルのFILTER列に、条件を通過した場合“PASS”、そうでない場合は “filterName”が記入される ※オプション DP:Approximate read depth; some reads may have been filtered SB:Strand Bias QD:Variant Confidence/Quality by Depth HRun:Largest Contiguous Homopolymer Run of Variant Allele In Either Direction Copyright © Amelieff Co. Ltd. All Rights Reserved. 16
17.
サ ン プ
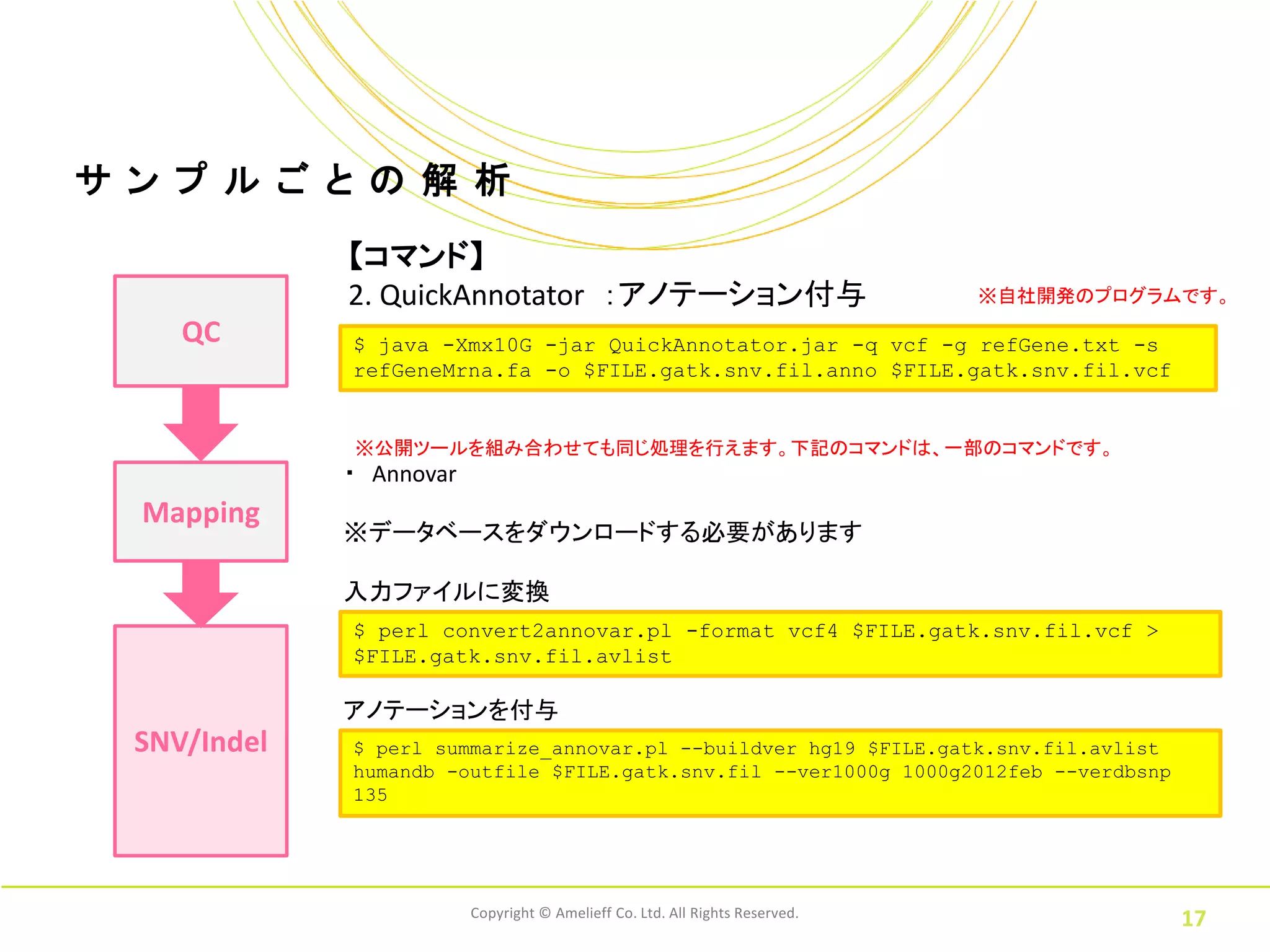
ル ご と の 解 析 【コマンド】 2. QuickAnnotator :アノテーション付与 ※自社開発のプログラムです。 QC $ java -Xmx10G -jar QuickAnnotator.jar -q vcf -g refGene.txt -s refGeneMrna.fa -o $FILE.gatk.snv.fil.anno $FILE.gatk.snv.fil.vcf ※公開ツールを組み合わせても同じ処理を行えます。下記のコマンドは、一部のコマンドです。 ・ Annovar Mapping ※データベースをダウンロードする必要があります 入力ファイルに変換 $ perl convert2annovar.pl -format vcf4 $FILE.gatk.snv.fil.vcf > $FILE.gatk.snv.fil.avlist アノテーションを付与 SNV/Indel $ perl summarize_annovar.pl --buildver hg19 $FILE.gatk.snv.fil.avlist humandb -outfile $FILE.gatk.snv.fil --ver1000g 1000g2012feb --verdbsnp 135 Copyright © Amelieff Co. Ltd. All Rights Reserved. 17
18.
サ ン プ
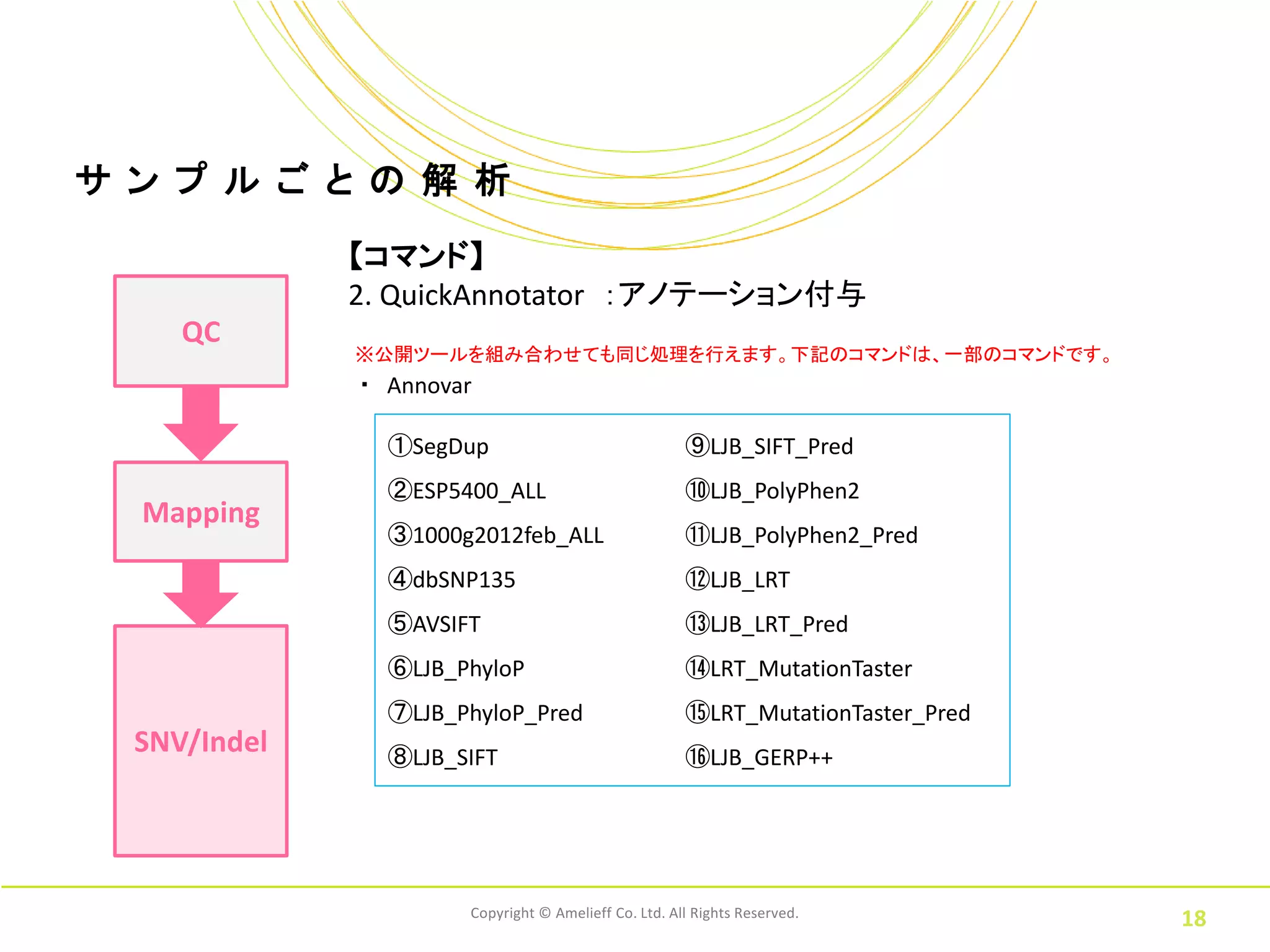
ル ご と の 解 析 【コマンド】 2. QuickAnnotator :アノテーション付与 QC ※公開ツールを組み合わせても同じ処理を行えます。下記のコマンドは、一部のコマンドです。 ・ Annovar ①SegDup ⑨LJB_SIFT_Pred ②ESP5400_ALL ⑩LJB_PolyPhen2 Mapping ③1000g2012feb_ALL ⑪LJB_PolyPhen2_Pred ④dbSNP135 ⑫LJB_LRT ⑤AVSIFT ⑬LJB_LRT_Pred ⑥LJB_PhyloP ⑭LRT_MutationTaster ⑦LJB_PhyloP_Pred ⑮LRT_MutationTaster_Pred SNV/Indel ⑧LJB_SIFT ⑯LJB_GERP++ Copyright © Amelieff Co. Ltd. All Rights Reserved. 18
19.
サ ン プ
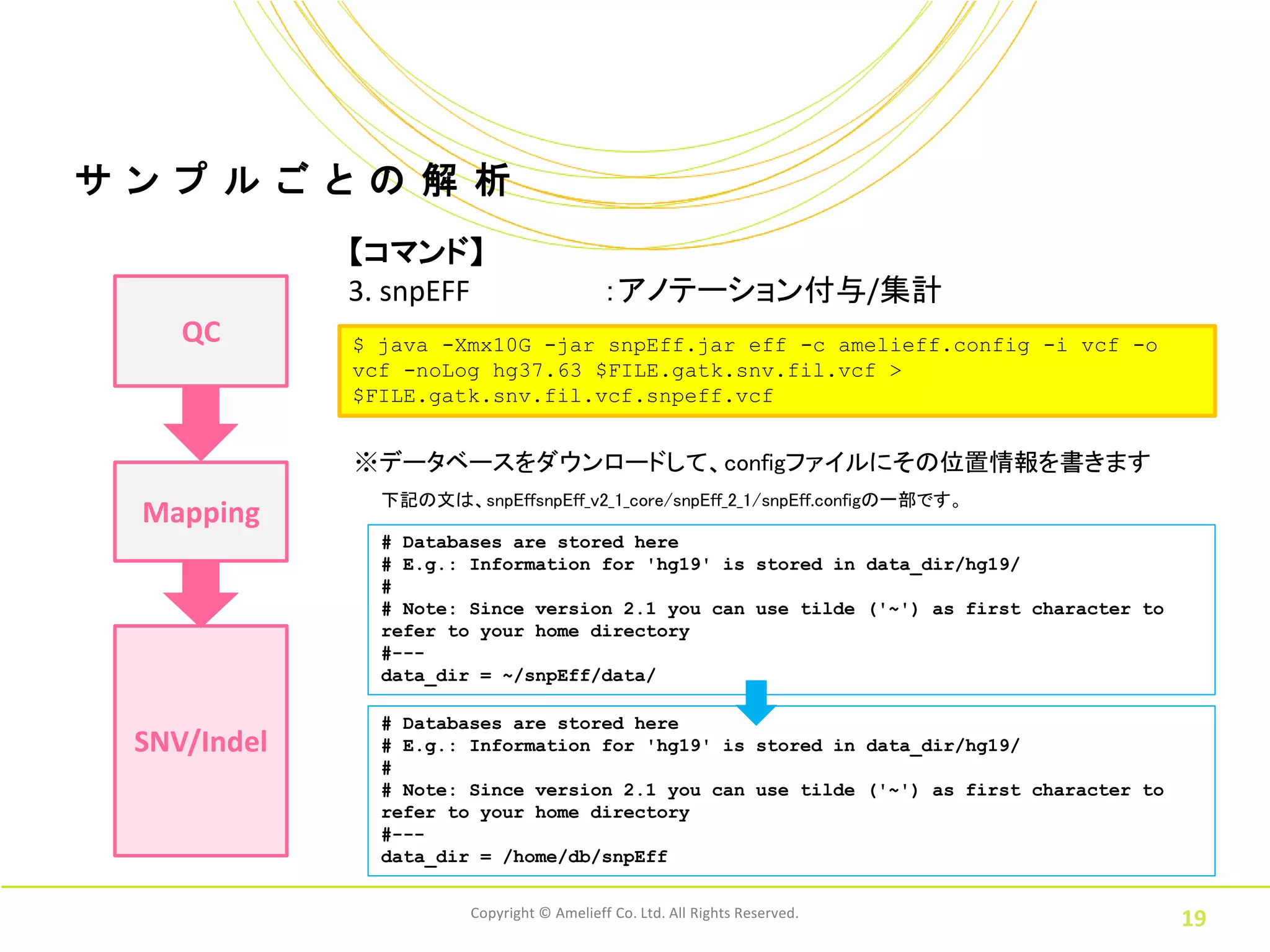
ル ご と の 解 析 【コマンド】 3. snpEFF :アノテーション付与/集計 QC $ java -Xmx10G -jar snpEff.jar eff -c amelieff.config -i vcf -o vcf -noLog hg37.63 $FILE.gatk.snv.fil.vcf > $FILE.gatk.snv.fil.vcf.snpeff.vcf ※データベースをダウンロードして、configファイルにその位置情報を書きます 下記の文は、snpEffsnpEff_v2_1_core/snpEff_2_1/snpEff.configの一部です。 Mapping # Databases are stored here # E.g.: Information for 'hg19' is stored in data_dir/hg19/ # # Note: Since version 2.1 you can use tilde ('~') as first character to refer to your home directory #--- data_dir = ~/snpEff/data/ # Databases are stored here SNV/Indel # E.g.: Information for 'hg19' is stored in data_dir/hg19/ # # Note: Since version 2.1 you can use tilde ('~') as first character to refer to your home directory #--- data_dir = /home/db/snpEff Copyright © Amelieff Co. Ltd. All Rights Reserved. 19
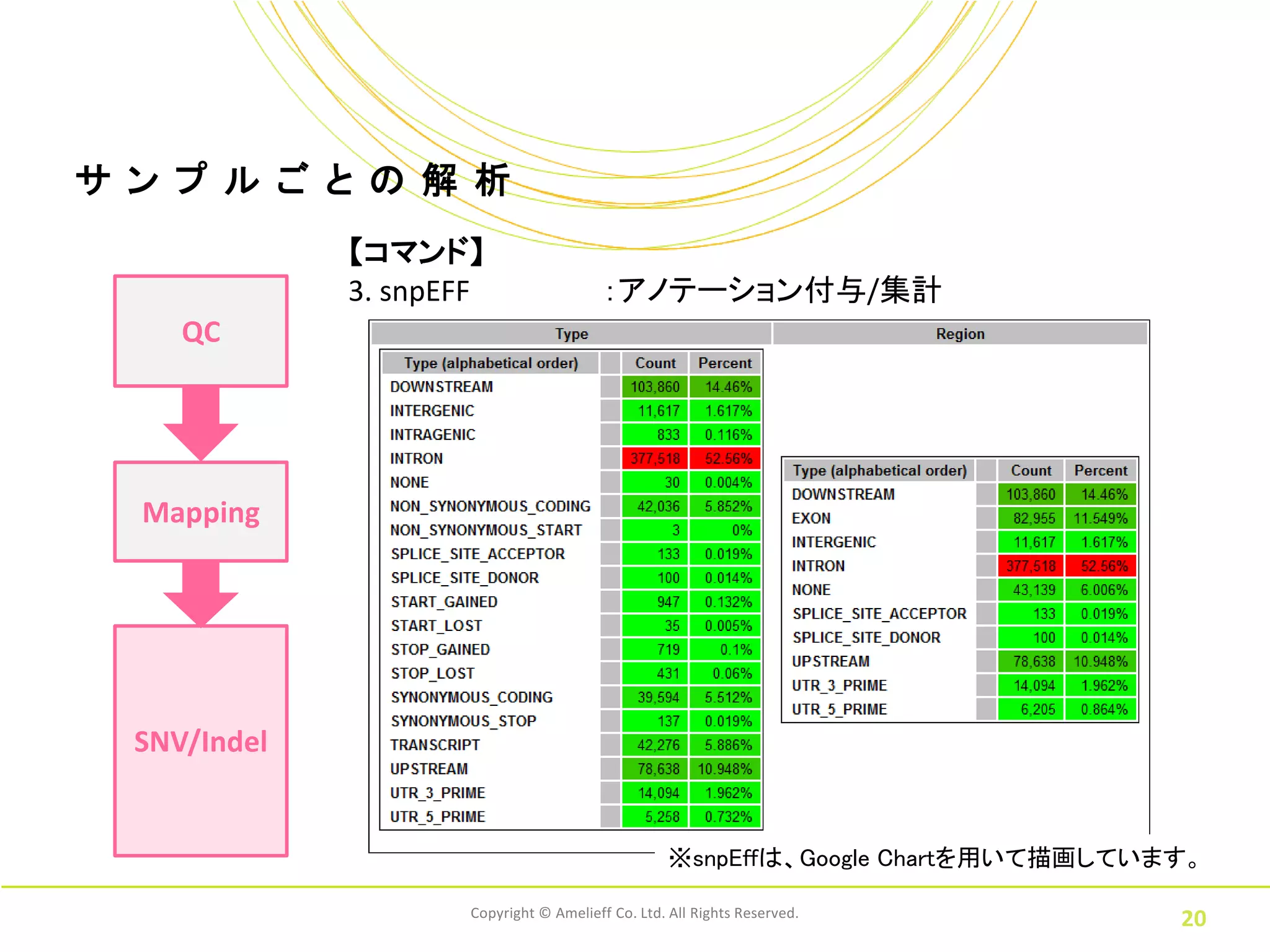
20.
サ ン プ
ル ご と の 解 析 【コマンド】 3. snpEFF :アノテーション付与/集計 QC Mapping SNV/Indel ※snpEffは、Google Chartを用いて描画しています。 Copyright © Amelieff Co. Ltd. All Rights Reserved. 20
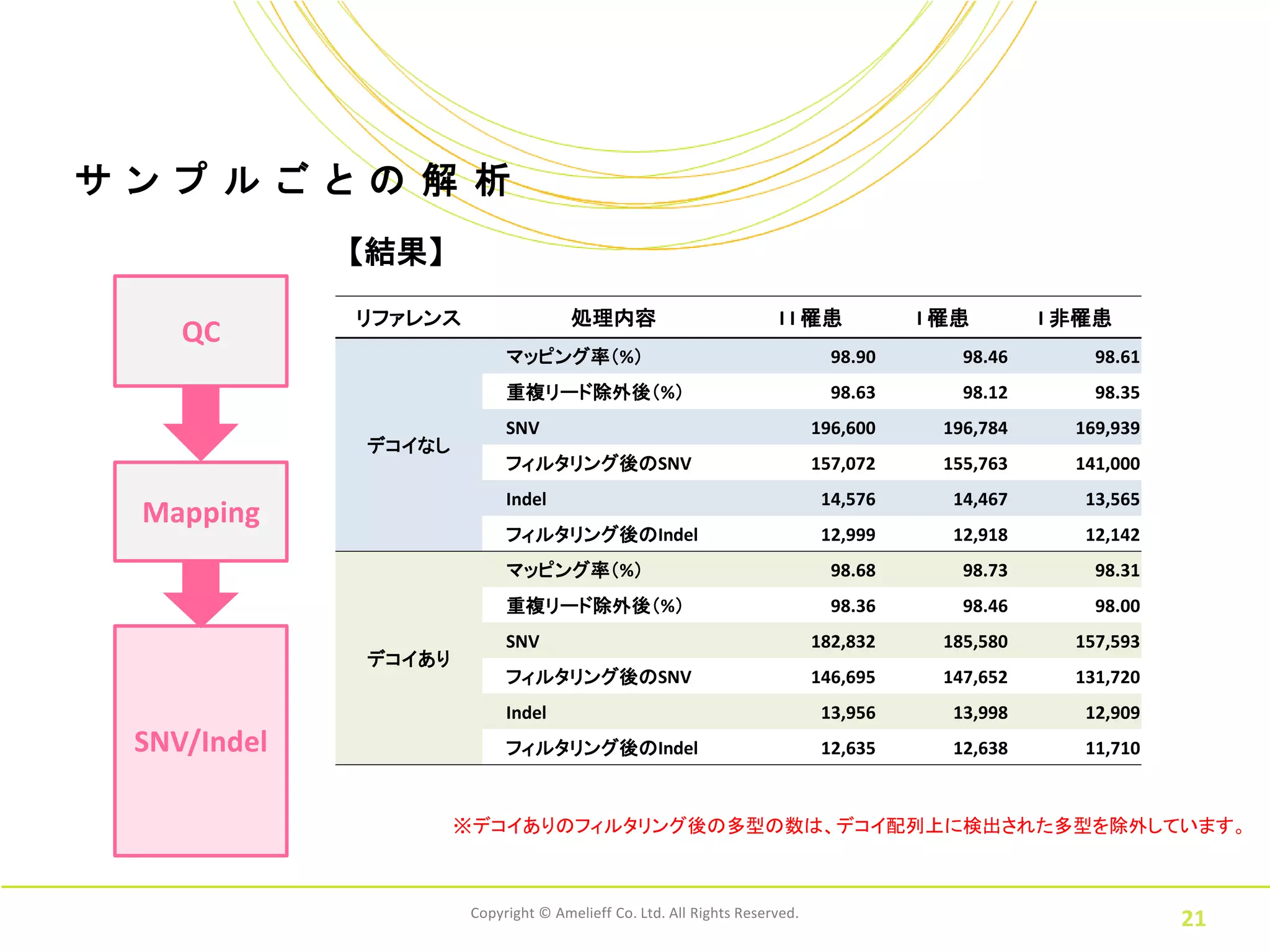
21.
サ ン プ
ル ご と の 解 析 【結果】 リファレンス 処理内容 I I 罹患 I 罹患 I 非罹患 QC マッピング率(%) 98.90 98.46 98.61 重複リード除外後(%) 98.63 98.12 98.35 SNV 196,600 196,784 169,939 デコイなし フィルタリング後のSNV 157,072 155,763 141,000 Indel 14,576 14,467 13,565 Mapping フィルタリング後のIndel 12,999 12,918 12,142 マッピング率(%) 98.68 98.73 98.31 重複リード除外後(%) 98.36 98.46 98.00 SNV 182,832 185,580 157,593 デコイあり フィルタリング後のSNV 146,695 147,652 131,720 Indel 13,956 13,998 12,909 SNV/Indel フィルタリング後のIndel 12,635 12,638 11,710 ※デコイありのフィルタリング後の多型の数は、デコイ配列上に検出された多型を除外しています。 Copyright © Amelieff Co. Ltd. All Rights Reserved. 21
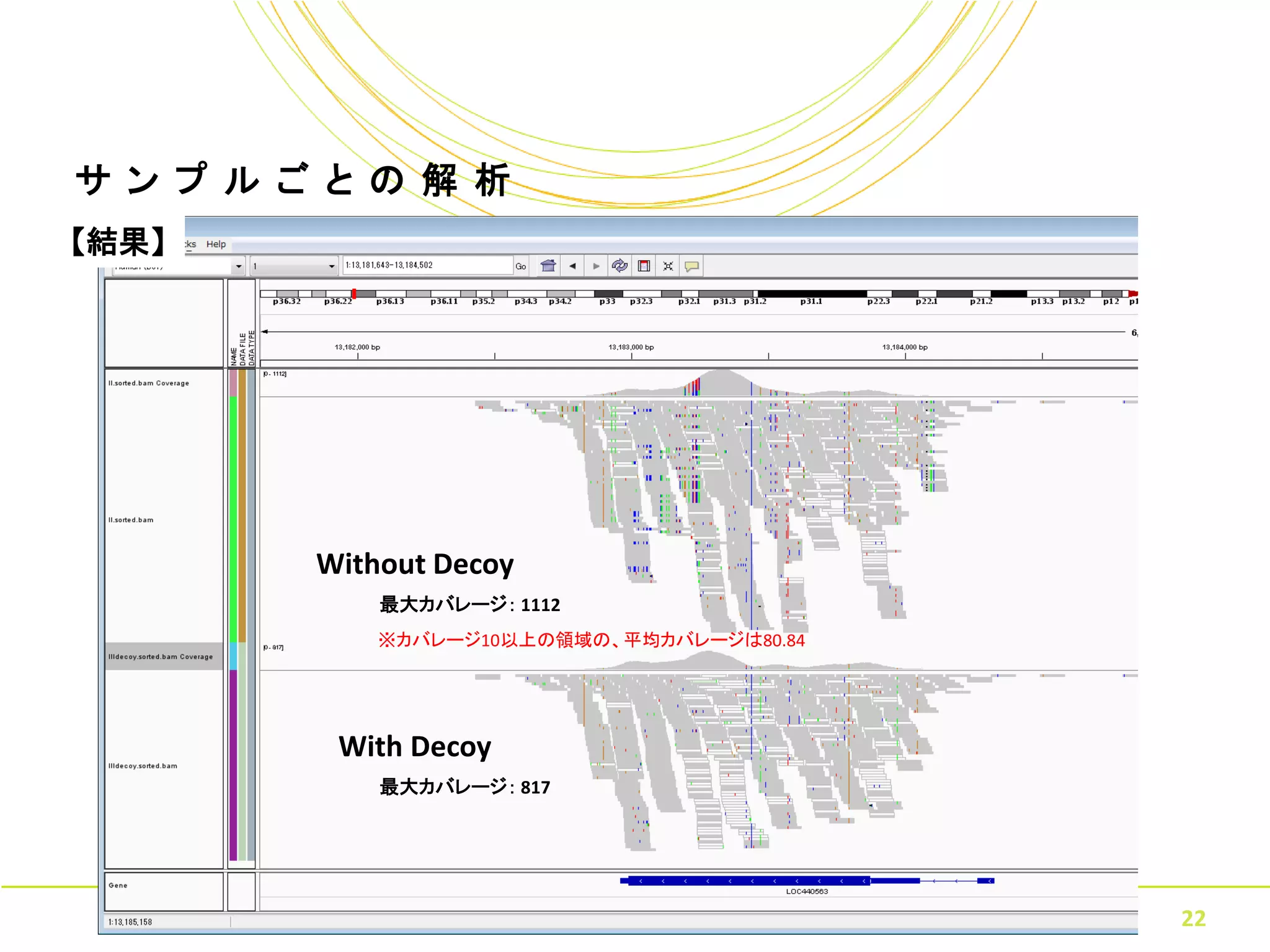
22.
サ ン プ
ル ご と の 解 析 【結果】 Without Decoy 最大カバレージ: 1112 ※カバレージ10以上の領域の、平均カバレージは80.84 With Decoy 最大カバレージ: 817 Copyright © Amelieff Co. Ltd. All Rights Reserved. 22
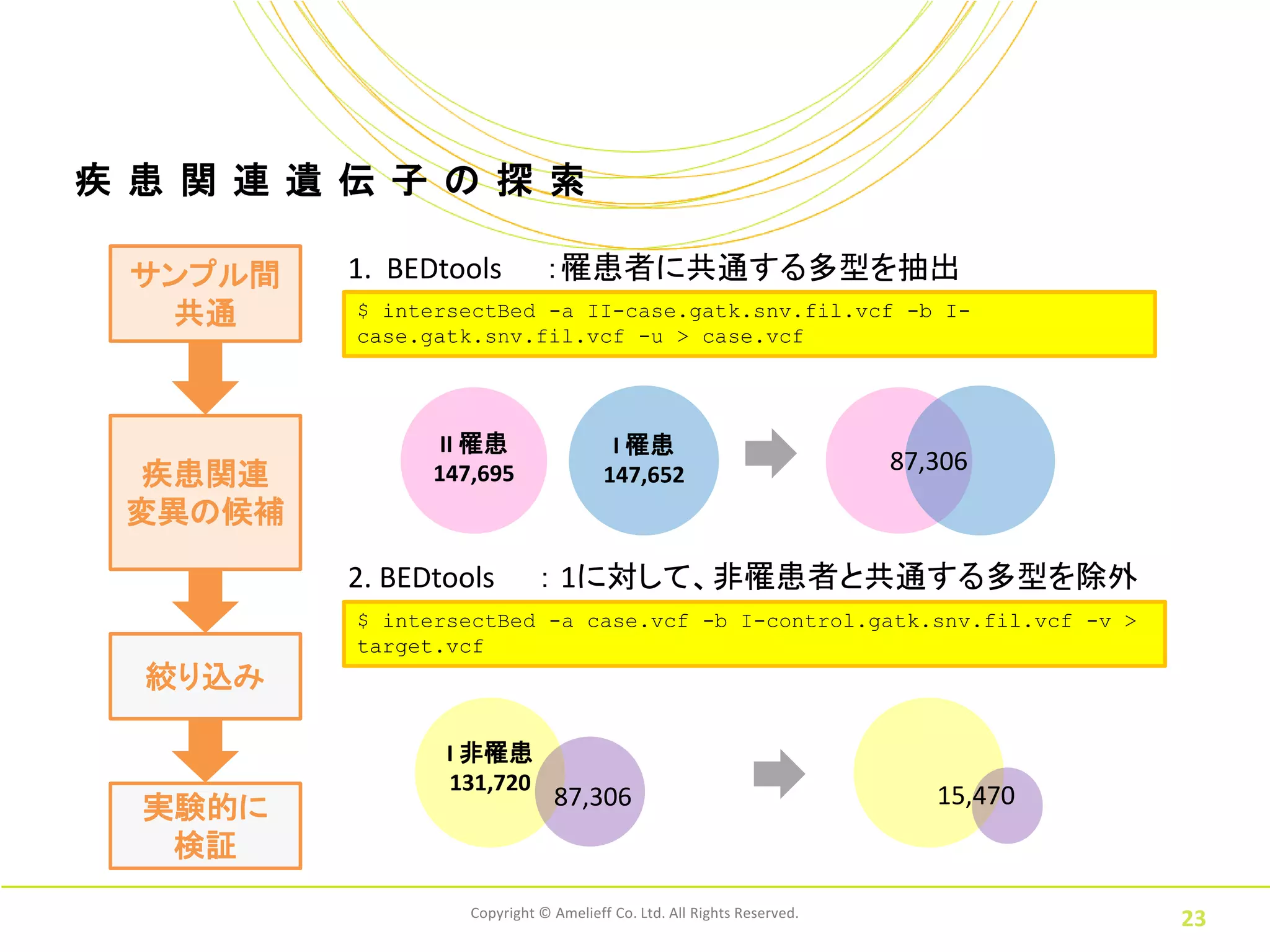
23.
疾 患 関
連 遺 伝 子 の 探 索 サンプル間 1. BEDtools :罹患者に共通する多型を抽出 共通 $ intersectBed -a II-case.gatk.snv.fil.vcf -b I- case.gatk.snv.fil.vcf -u > case.vcf II 罹患 I 罹患 87,306 疾患関連 147,695 147,652 変異の候補 2. BEDtools : 1に対して、非罹患者と共通する多型を除外 $ intersectBed -a case.vcf -b I-control.gatk.snv.fil.vcf -v > target.vcf 絞り込み I 非罹患 131,720 実験的に 87,306 15,470 検証 Copyright © Amelieff Co. Ltd. All Rights Reserved. 23
24.
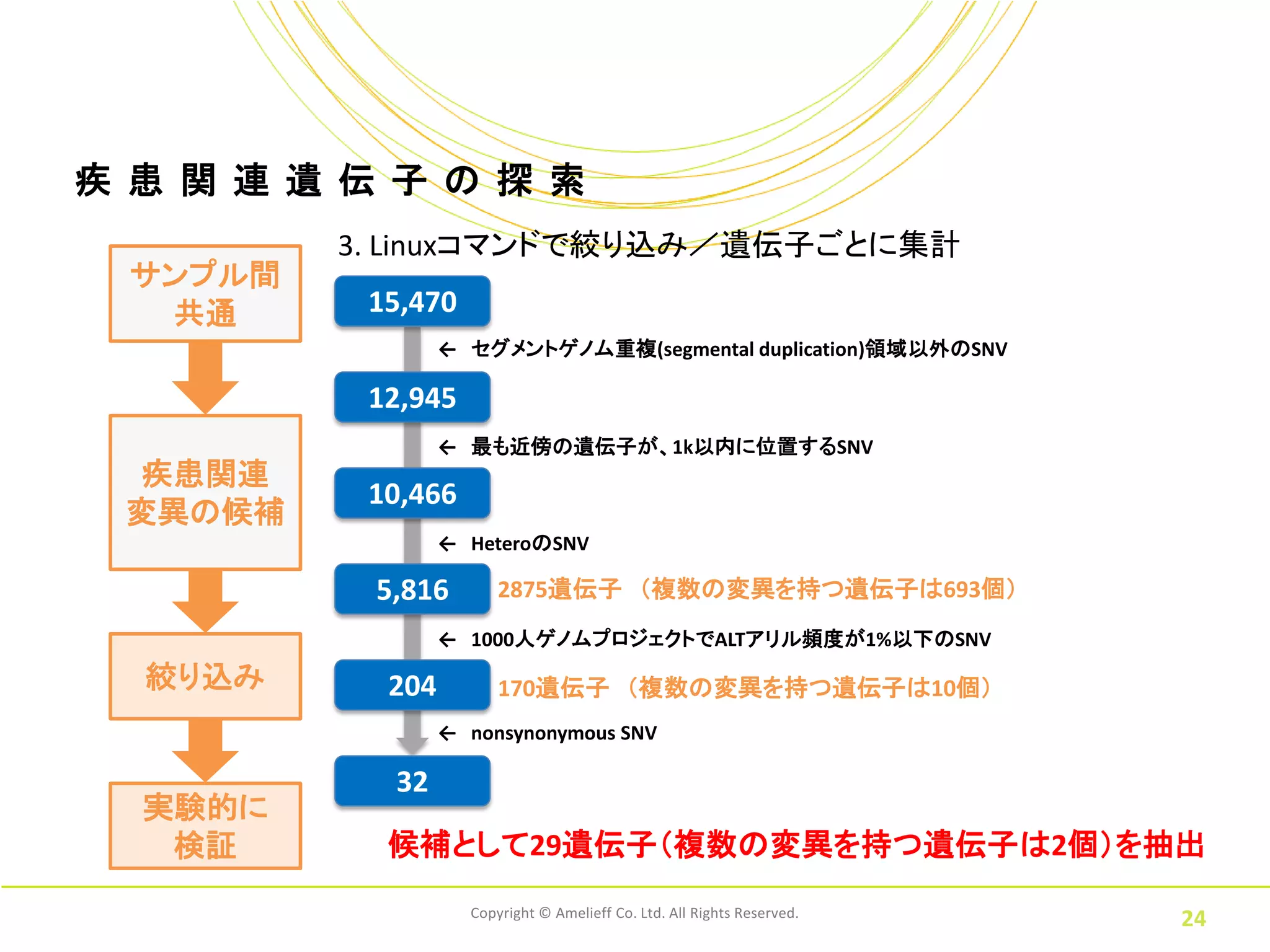
疾 患 関
連 遺 伝 子 の 探 索 3. Linuxコマンドで絞り込み/遺伝子ごとに集計 サンプル間 共通 15,470 ← セグメントゲノム重複(segmental duplication)領域以外のSNV 12,945 ← 最も近傍の遺伝子が、1k以内に位置するSNV 疾患関連 10,466 変異の候補 ← HeteroのSNV 5,816 2875遺伝子 (複数の変異を持つ遺伝子は693個) ← 1000人ゲノムプロジェクトでALTアリル頻度が1%以下のSNV 絞り込み 204 170遺伝子 (複数の変異を持つ遺伝子は10個) ← nonsynonymous SNV 32 実験的に 検証 候補として29遺伝子(複数の変異を持つ遺伝子は2個)を抽出 Copyright © Amelieff Co. Ltd. All Rights Reserved. 24
25.
NGSデータ解析サーバー販売中!!
データ解析パイプラインおよびリファレンスゲノム情報がプリインストールされています。 今回ご紹介した解析は、下記のサーバーで実施しました。サンプルあたりの実行時間は約8時間です。 OS CentOS 6 64bit CPU Intel Core i7-3930K [3.20GHz/6Core] メモリ 64GB SSD 64GBz(OS用) HDD 2TB x 4台(RAID10)(データ用) bwa, SAMtools, FastQC, BEDtools, ソフトウェア 自社開発ソフトなど Copyright © Amelieff Co. Ltd. All Rights Reserved. 25

![サ ン プ ル ご と の 解 析
【コマンド】
1. QCleaner : Fastqファイルをフィルタリング ※自社開発のプログラムです。
QC $ perl qcleaner.pl --i1 $FILE1.fastq --i2 $FILE2.fastq --o1
$FILE1.clean.fastq --o2 $FILE2.clean.fastq --qp 20,80 --n 10 --
trim 20 --length 20 --log $FILE.qc.log
FASTQ形式にマッチするかチェック
データクオリティチェック(FastQC)
Mapping
Illumina CASAVA filter [Y] を除去
クオリティ20未満が80%以上のリードを除去
クオリティ20未満の末端をトリム
未知の塩基(N)が多いリード除去
SNV/Indel 配列長が短いリード除去
片側のみのリードを除外
データクオリティチェック(FastQC)
Copyright © Amelieff Co. Ltd. All Rights Reserved.
6](https://image.slidesharecdn.com/120707slideshare-120716203841-phpapp02/75/Exome-6-2048.jpg)
![サ ン プ ル ご と の 解 析
【コマンド】
1. BWA : マッピング
QC ・ マッピング/SAMをBAMに変換/ソート
$ bwa aln -t 6 genome_hs37d5cs.fa $FILE1.clean.fastq -f
$FILE1.sai
$ bwa sampe -r "@RG¥tID:$FILE¥tSM:$FILE¥tPL:Illumina" -n 3
Mapping -N 10 -a 500 genome_hs37d5cs.fa $FILE1.sai $FILE2.sai
$FILE1.clean.fastq $FILE2.clean.fastq | samtools view -Sb -
| samtools sort - $FILE.sorted
※オプション
-a INT maximum insert size [500]
-n INT maximum hits to output for paired reads [3]
-N INT maximum hits to output for discordant pairs [10]
SNV/Indel ※RG(read groups)
platform (PL) および sample (SM)が必要
PLの例:454, LS454, Illumina, Solid, ABI_Solid, CG (all case-insensitive)
解析ツールGATKに入力するBAMファイルに、RGタグの記述がないとエラーが出る
Copyright © Amelieff Co. Ltd. All Rights Reserved.
10](https://image.slidesharecdn.com/120707slideshare-120716203841-phpapp02/75/Exome-10-2048.jpg)
![NGSデータ解析サーバー販売中!!
データ解析パイプラインおよびリファレンスゲノム情報がプリインストールされています。
今回ご紹介した解析は、下記のサーバーで実施しました。サンプルあたりの実行時間は約8時間です。
OS CentOS 6 64bit
CPU Intel Core i7-3930K [3.20GHz/6Core]
メモリ 64GB
SSD 64GBz(OS用)
HDD 2TB x 4台(RAID10)(データ用)
bwa, SAMtools, FastQC, BEDtools,
ソフトウェア
自社開発ソフトなど
Copyright © Amelieff Co. Ltd. All Rights Reserved.
25](https://image.slidesharecdn.com/120707slideshare-120716203841-phpapp02/75/Exome-25-2048.jpg)





![[DDBJing33] ゲノムワイド多型を利用した遺伝解析の実際](https://cdn.slidesharecdn.com/ss_thumbnails/33ddbjingshirasawa-151120004737-lva1-app6891-thumbnail.jpg?width=640&height=640&fit=bounds)
![[DL輪読会]FOTS: Fast Oriented Text Spotting with a Unified Network](https://cdn.slidesharecdn.com/ss_thumbnails/20181012yokota-181012004624-thumbnail.jpg?width=640&height=640&fit=bounds)











![[DL輪読会]Flow-based Deep Generative Models](https://cdn.slidesharecdn.com/ss_thumbnails/20190307-190328024744-thumbnail.jpg?width=640&height=640&fit=bounds)
![[DL輪読会]data2vec: A General Framework for Self-supervised Learning in Speech,...](https://cdn.slidesharecdn.com/ss_thumbnails/220204nonakadl1-220204025334-thumbnail.jpg?width=640&height=640&fit=bounds)


![[DL輪読会]Learning convolutional neural networks for graphs](https://cdn.slidesharecdn.com/ss_thumbnails/learningconvolutionalneuralnetworksforgraphs-170222032121-thumbnail.jpg?width=640&height=640&fit=bounds)
























