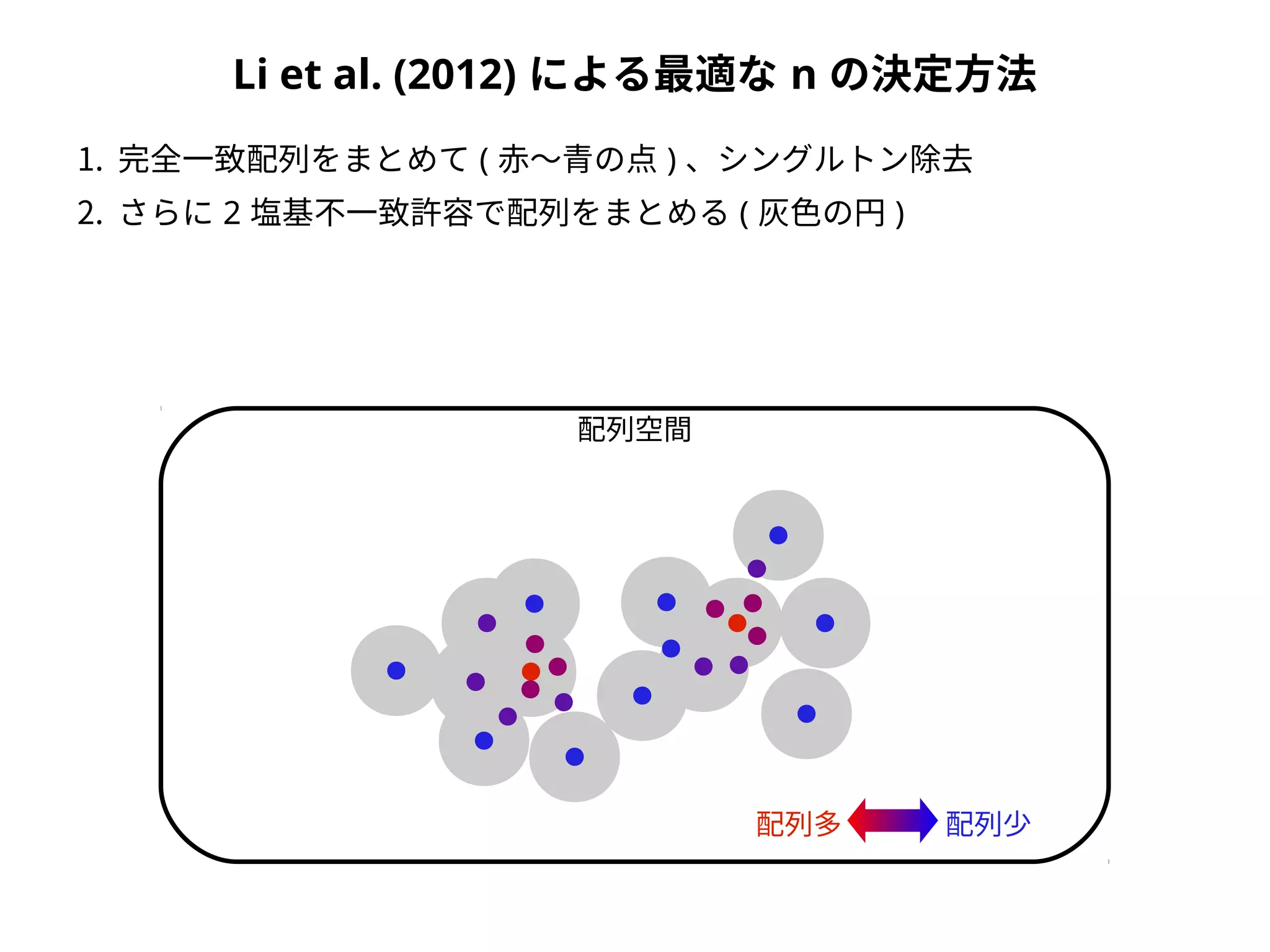
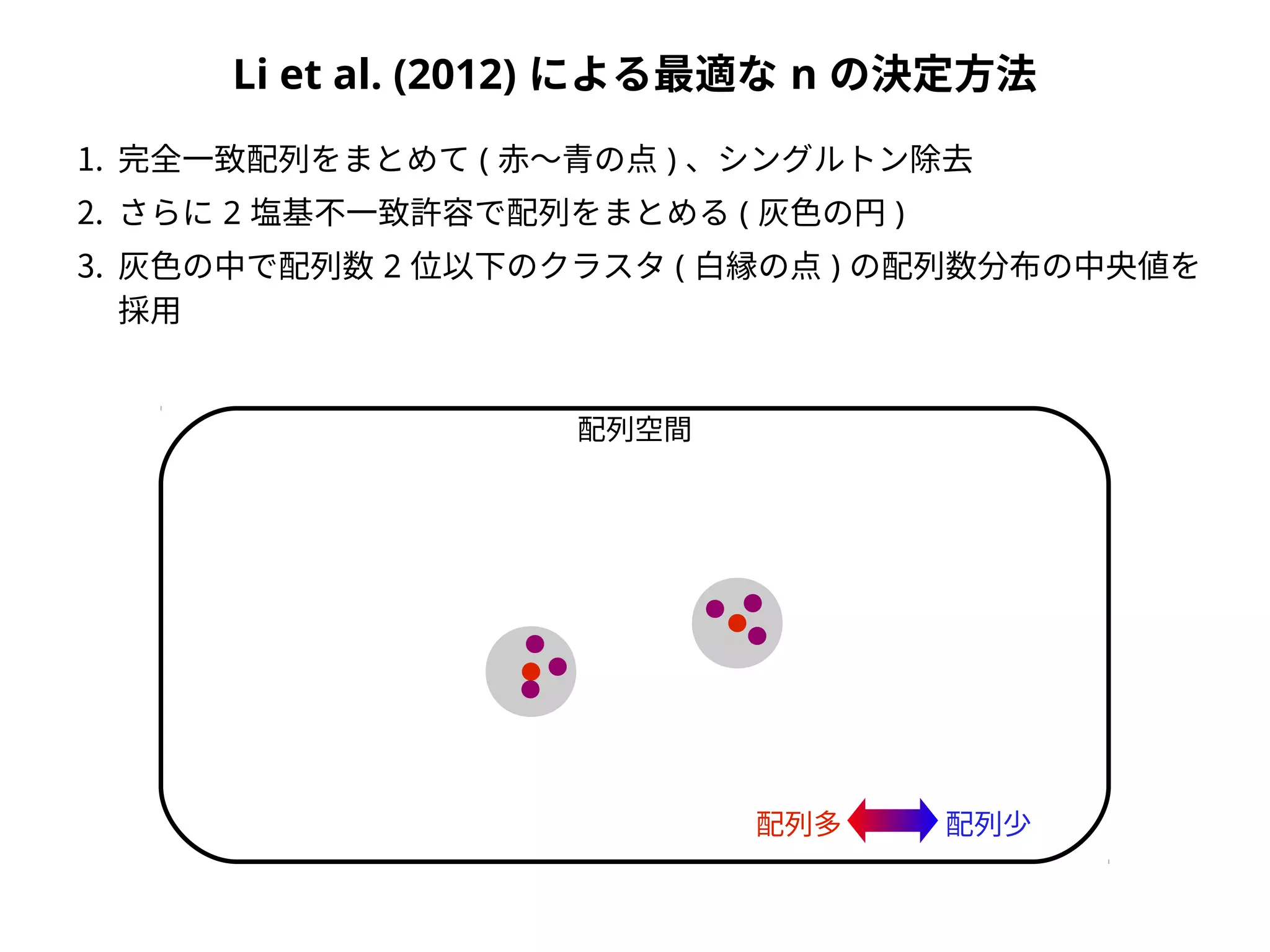
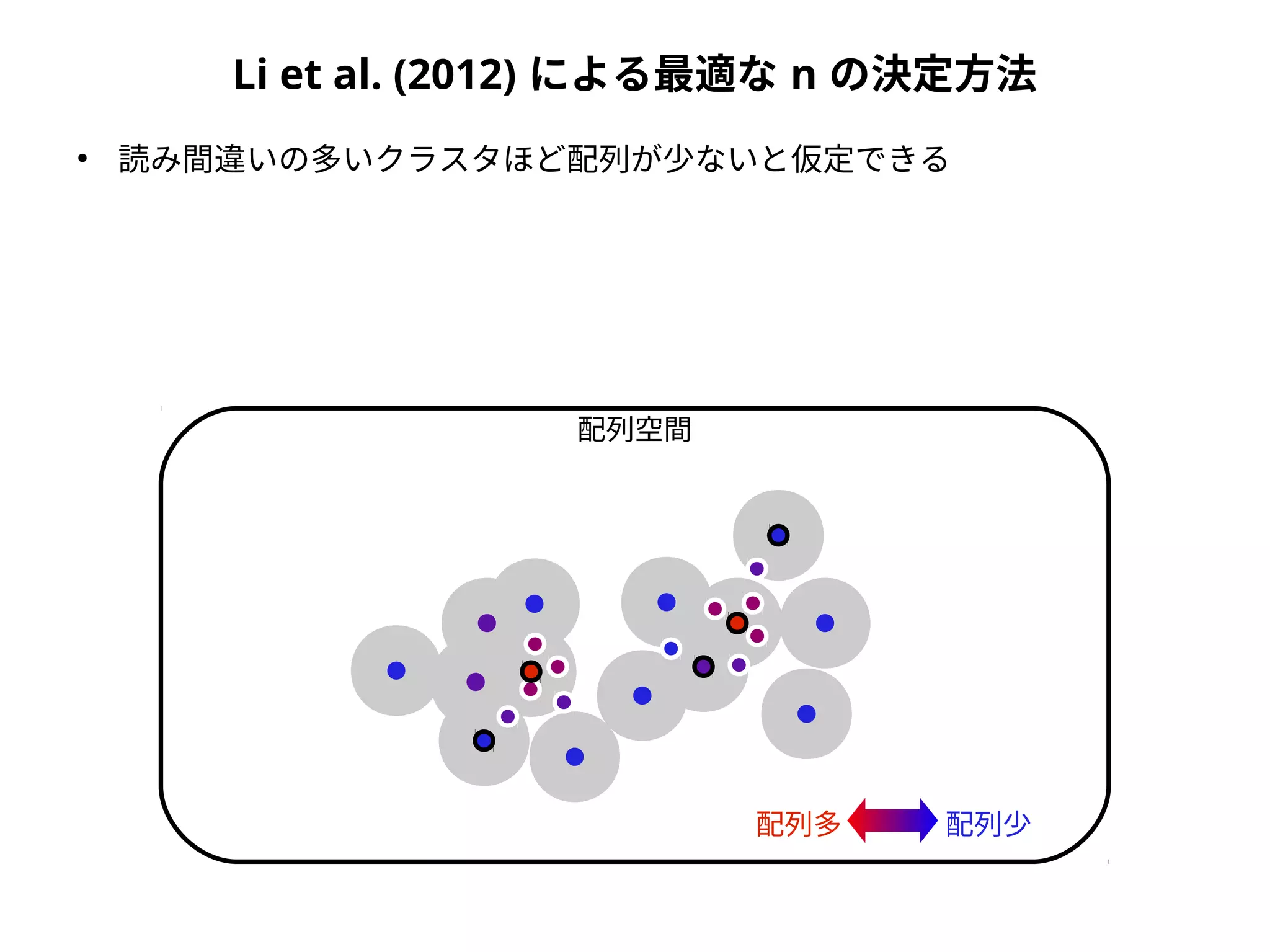
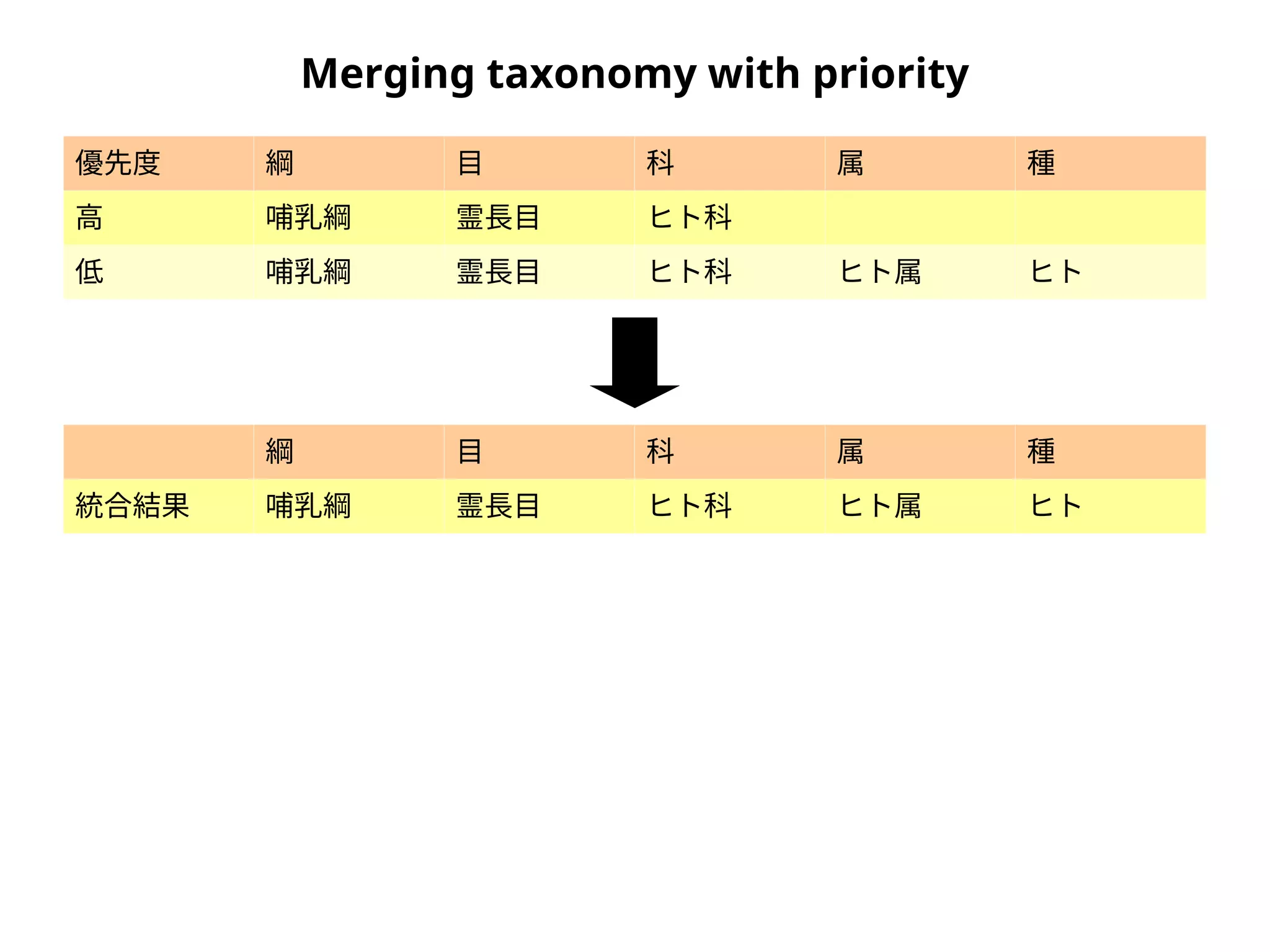
Taxonomic assignment
OTU AOTU B OTU C
Sample 1 2 3 0
Sample 2 1 1 3
family genus species
OTU A Foo Hoge Hoge fuga
OTU B Foo Piyo Piyo piyo
OTU C Bar Baz Baz qux
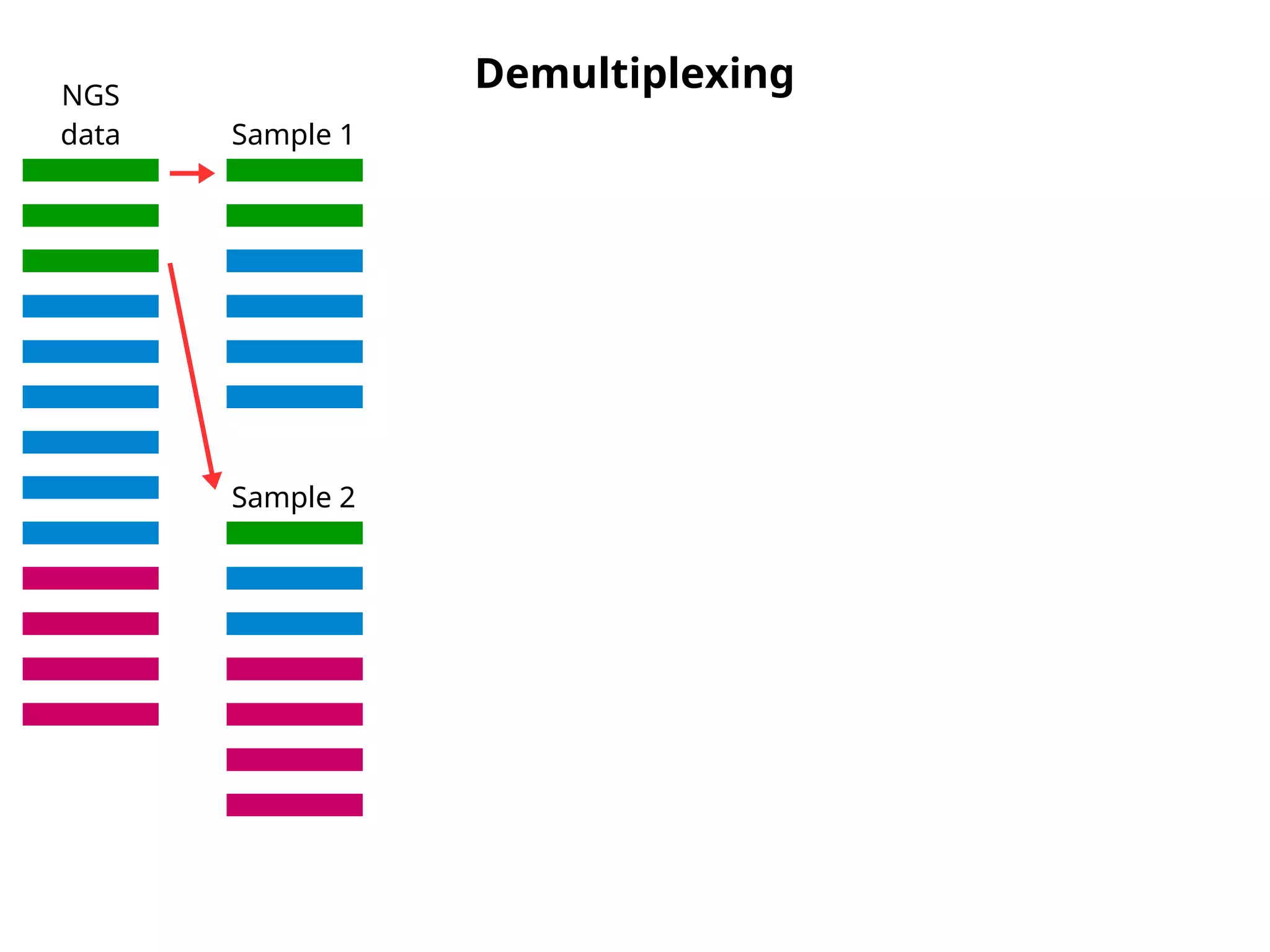
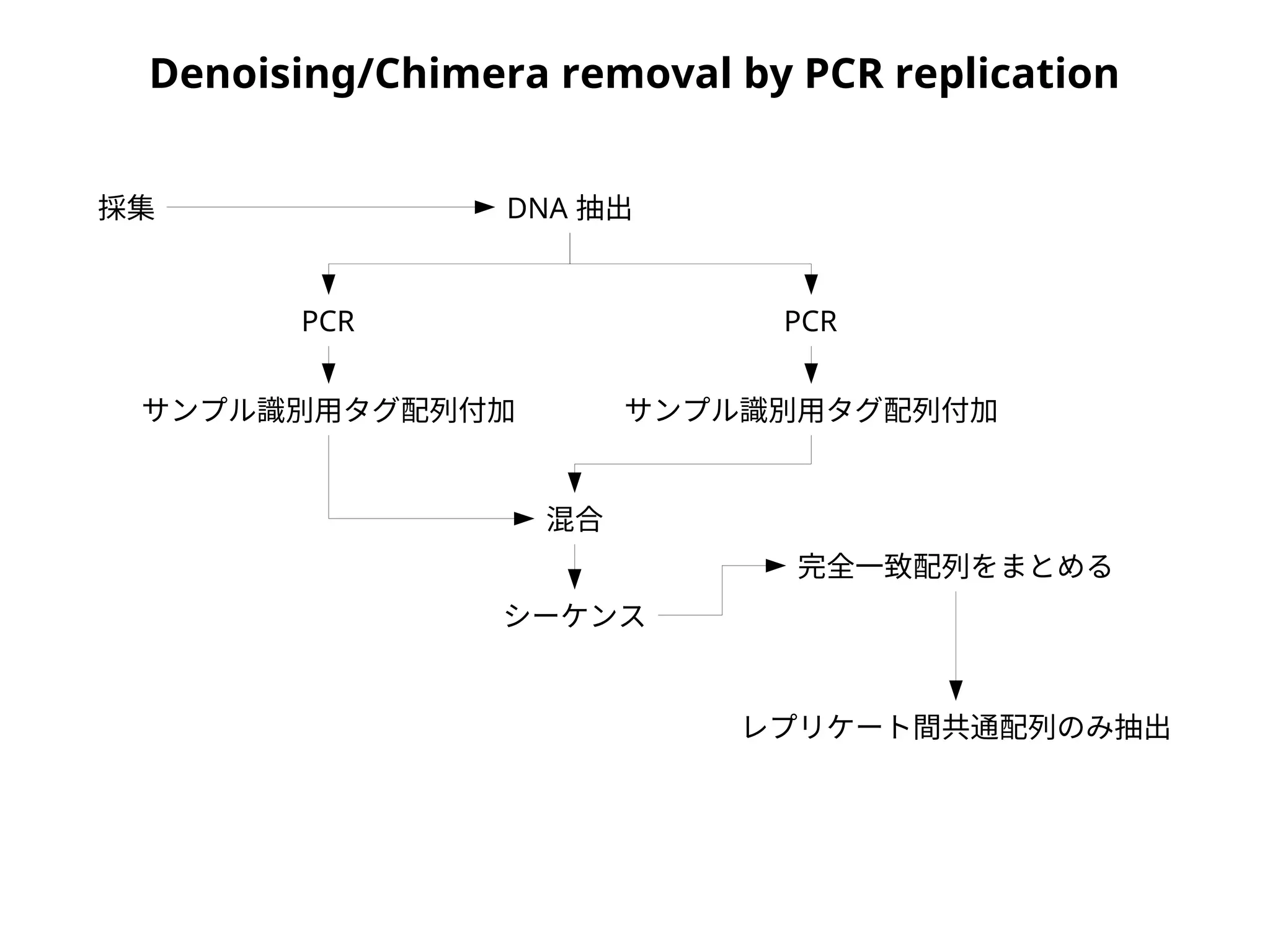
NGS
data Sample 1
Sample 2
Sample 1
Sample 2
OTUs
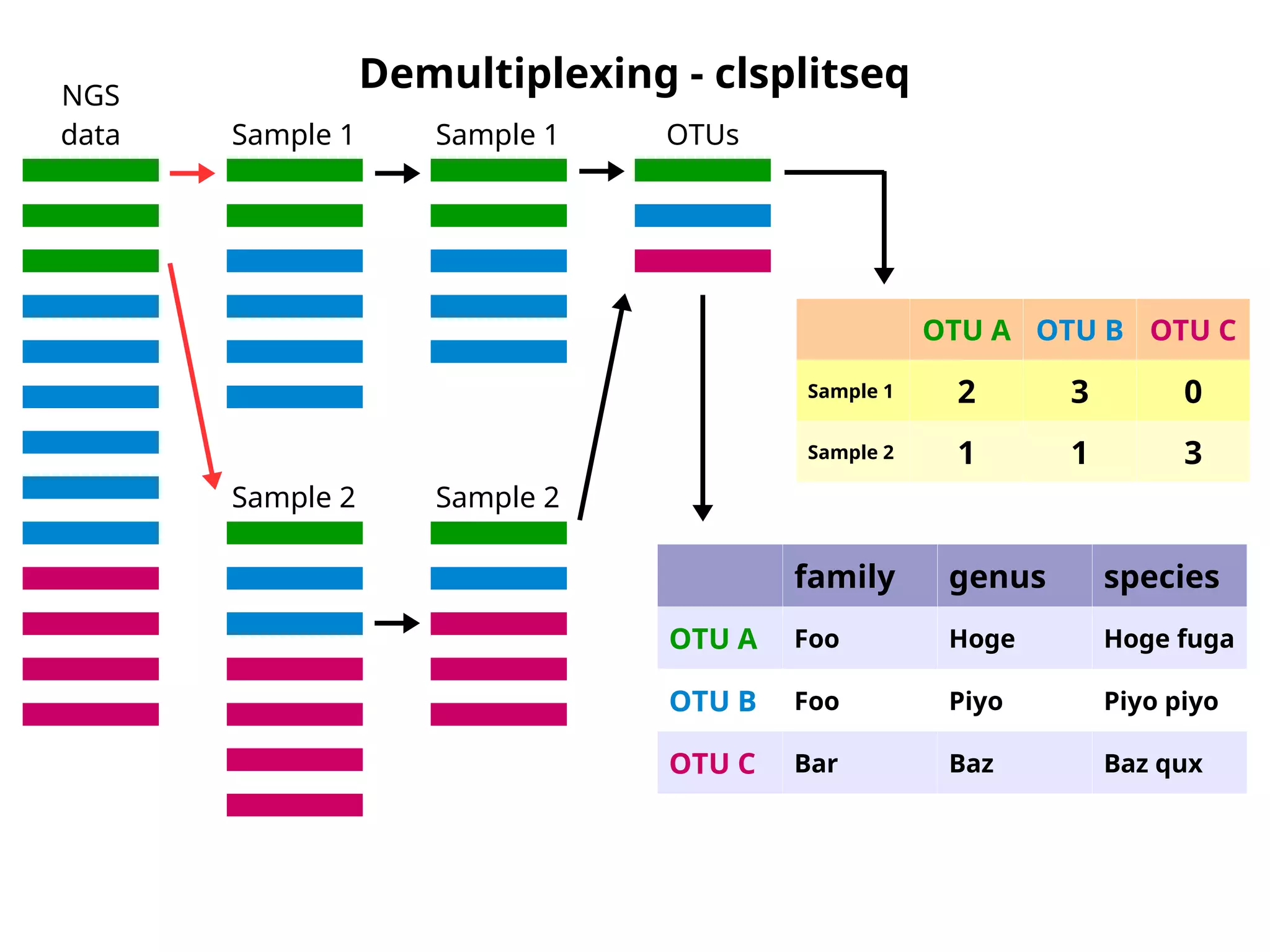
Demultiplexing - clsplitseq
OTUA OTU B OTU C
Sample 1 2 3 0
Sample 2 1 1 3
family genus species
OTU A Foo Hoge Hoge fuga
OTU B Foo Piyo Piyo piyo
OTU C Bar Baz Baz qux
NGS
data Sample 1
Sample 2
Sample 1
Sample 2
OTUs
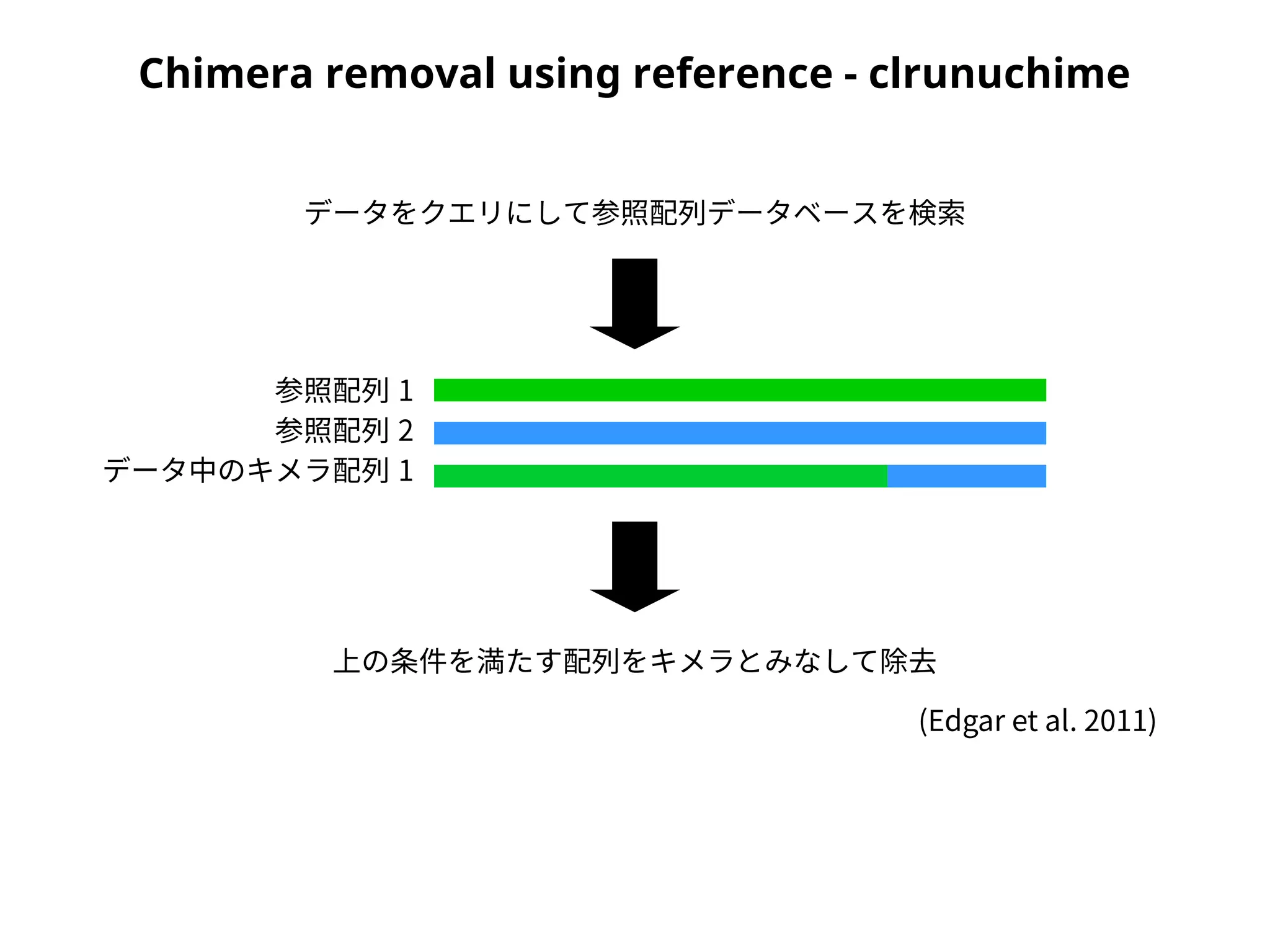
Denoising/Chimera removal -clcleanseqv
OTU A OTU B OTU C
Sample 1 2 3 0
Sample 2 1 1 3
family genus species
OTU A Foo Hoge Hoge fuga
OTU B Foo Piyo Piyo piyo
OTU C Bar Baz Baz qux
NGS
data Sample 1
Sample 2
Sample 1
Sample 2
OTUs
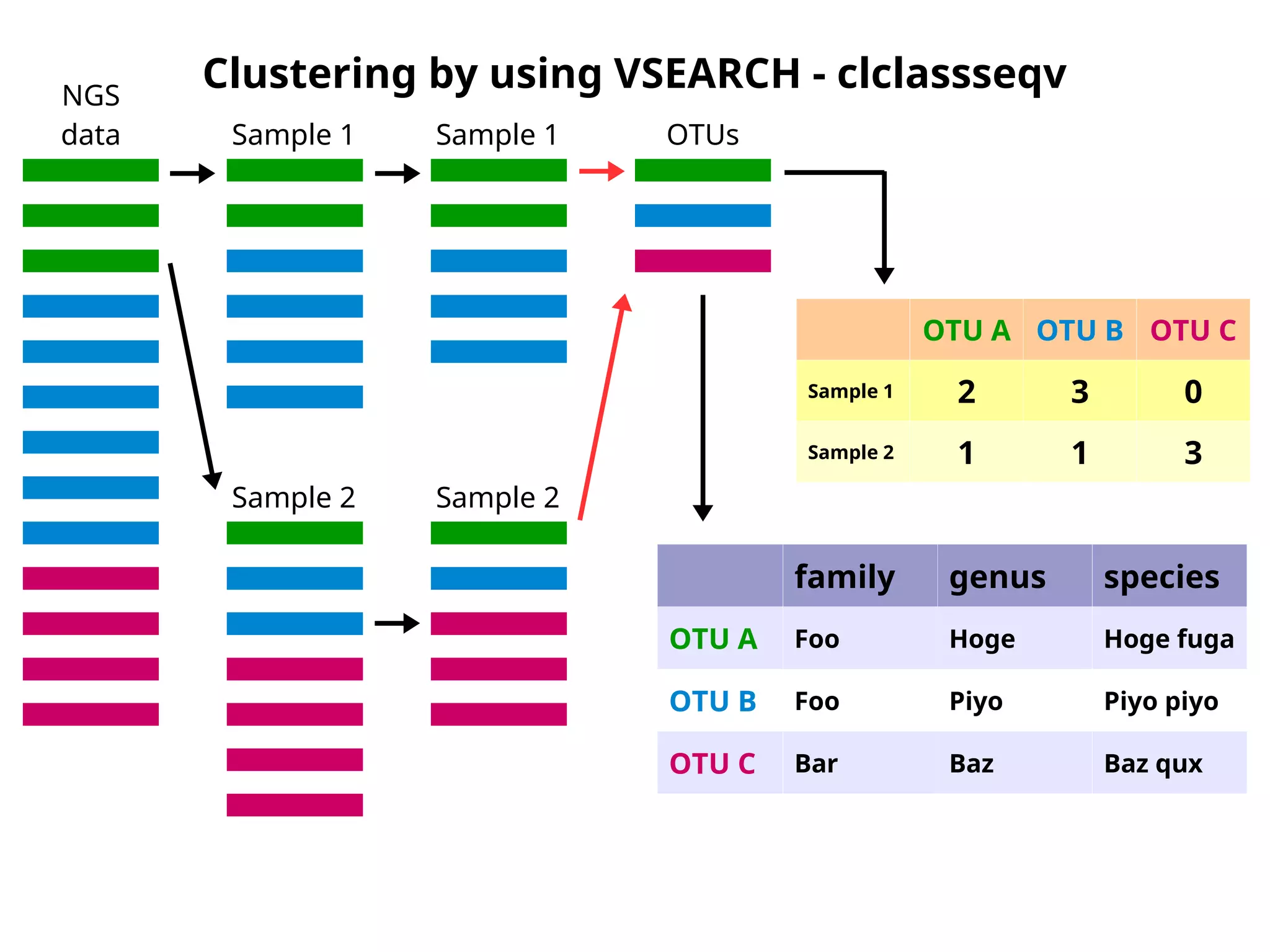
Clustering by usingVSEARCH - clclassseqv
OTU A OTU B OTU C
Sample 1 2 3 0
Sample 2 1 1 3
family genus species
OTU A Foo Hoge Hoge fuga
OTU B Foo Piyo Piyo piyo
OTU C Bar Baz Baz qux
NGS
data Sample 1
Sample 2
Sample 1
Sample 2
OTUs
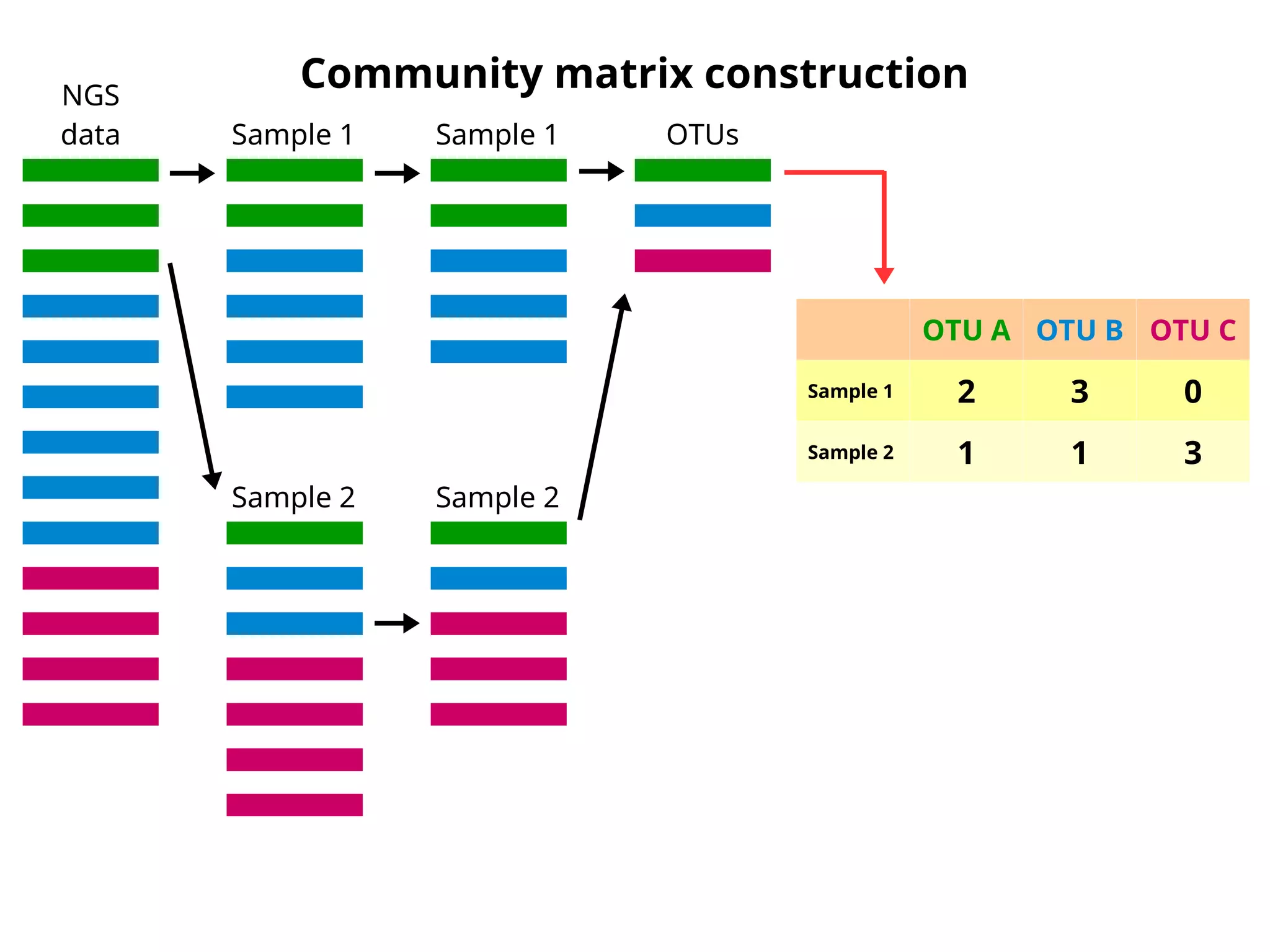
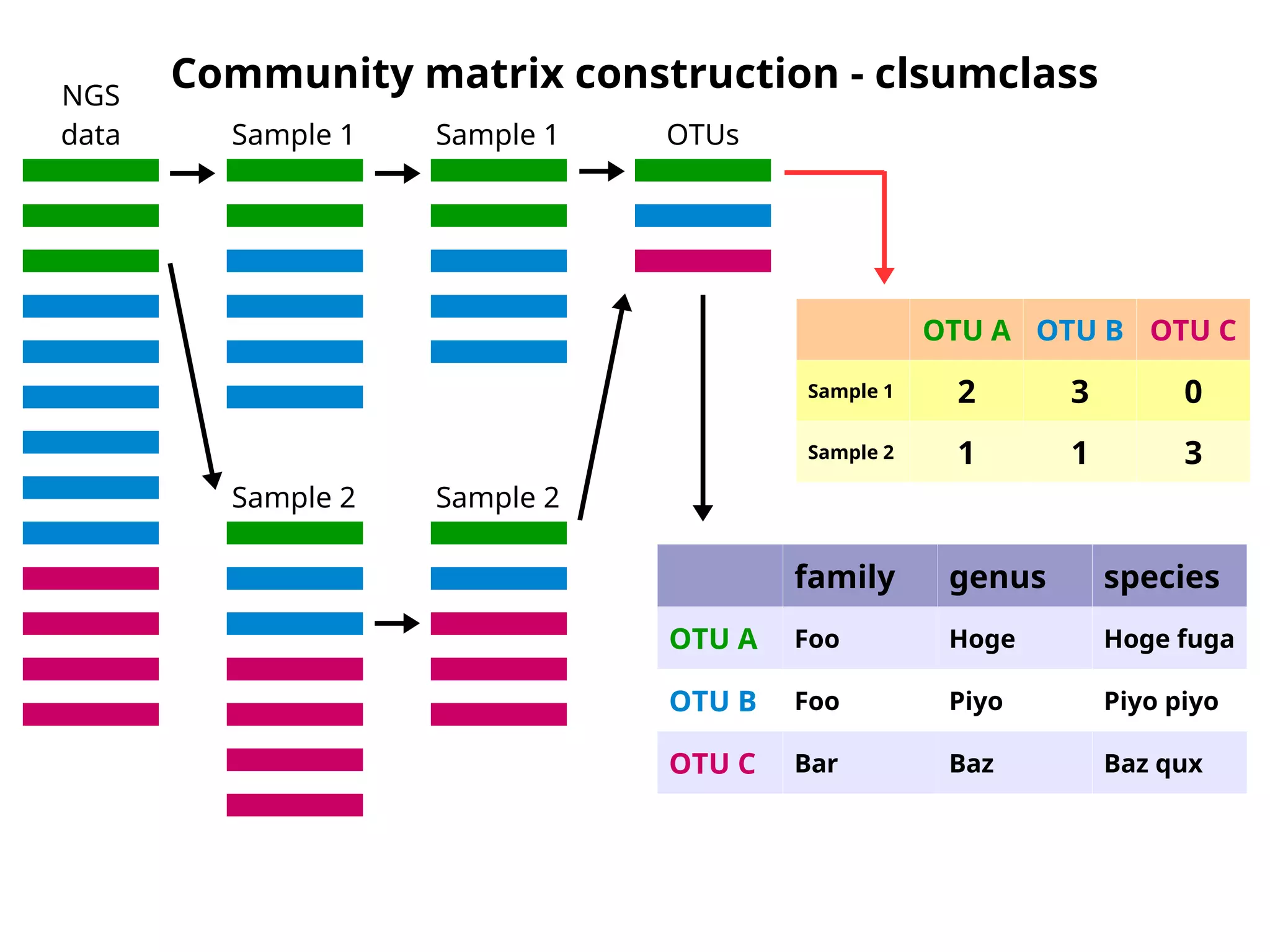
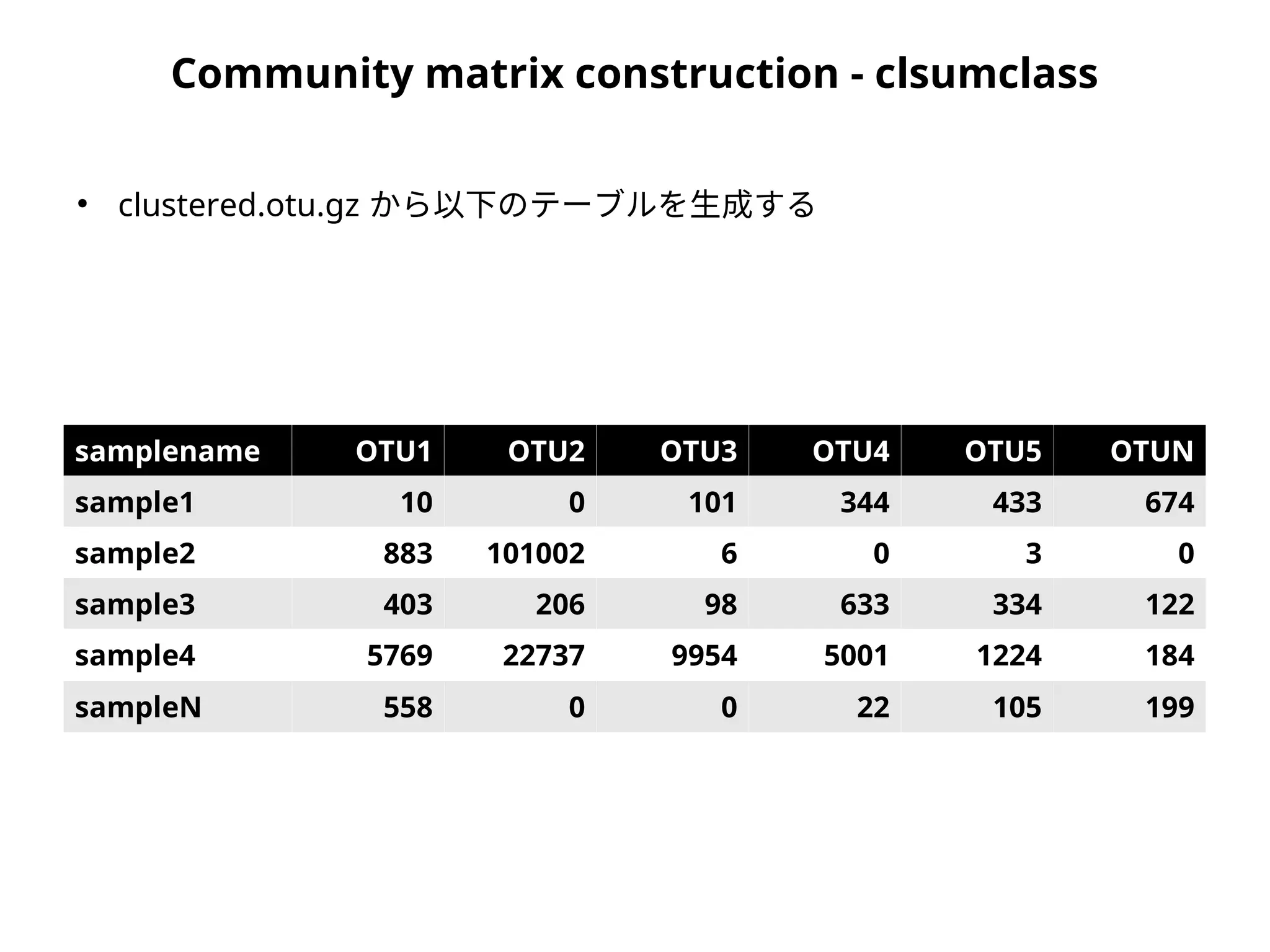
Community matrix construction- clsumclass
OTU A OTU B OTU C
Sample 1 2 3 0
Sample 2 1 1 3
family genus species
OTU A Foo Hoge Hoge fuga
OTU B Foo Piyo Piyo piyo
OTU C Bar Baz Baz qux
NGS
data Sample 1
Sample 2
Sample 1
Sample 2
OTUs
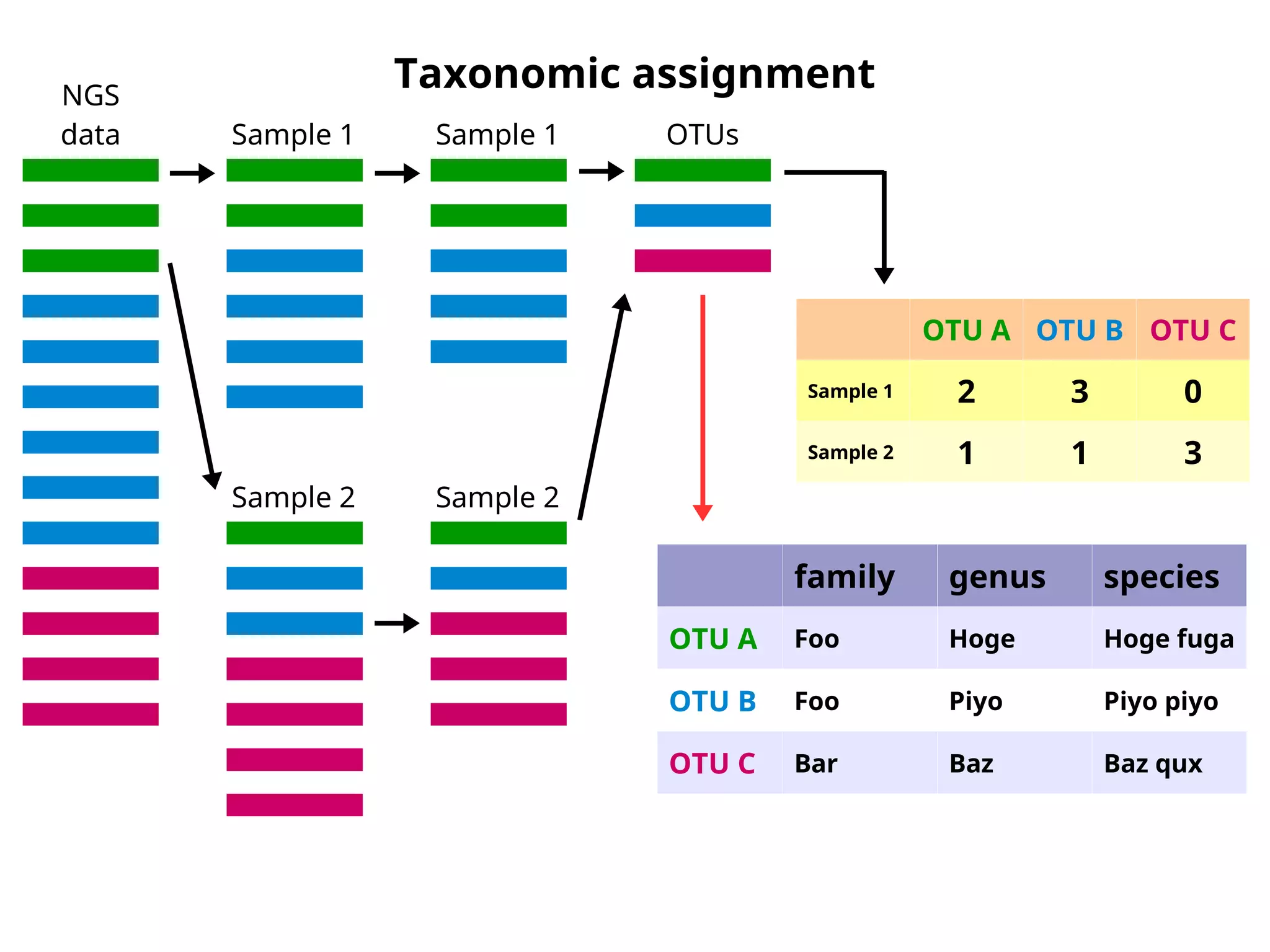
Taxonomic assignment -clidentseq/classigntax
OTU A OTU B OTU C
Sample 1 2 3 0
Sample 2 1 1 3
family genus species
OTU A Foo Hoge Hoge fuga
OTU B Foo Piyo Piyo piyo
OTU C Bar Baz Baz qux
NGS
data Sample 1
Sample 2
Sample 1
Sample 2
OTUs
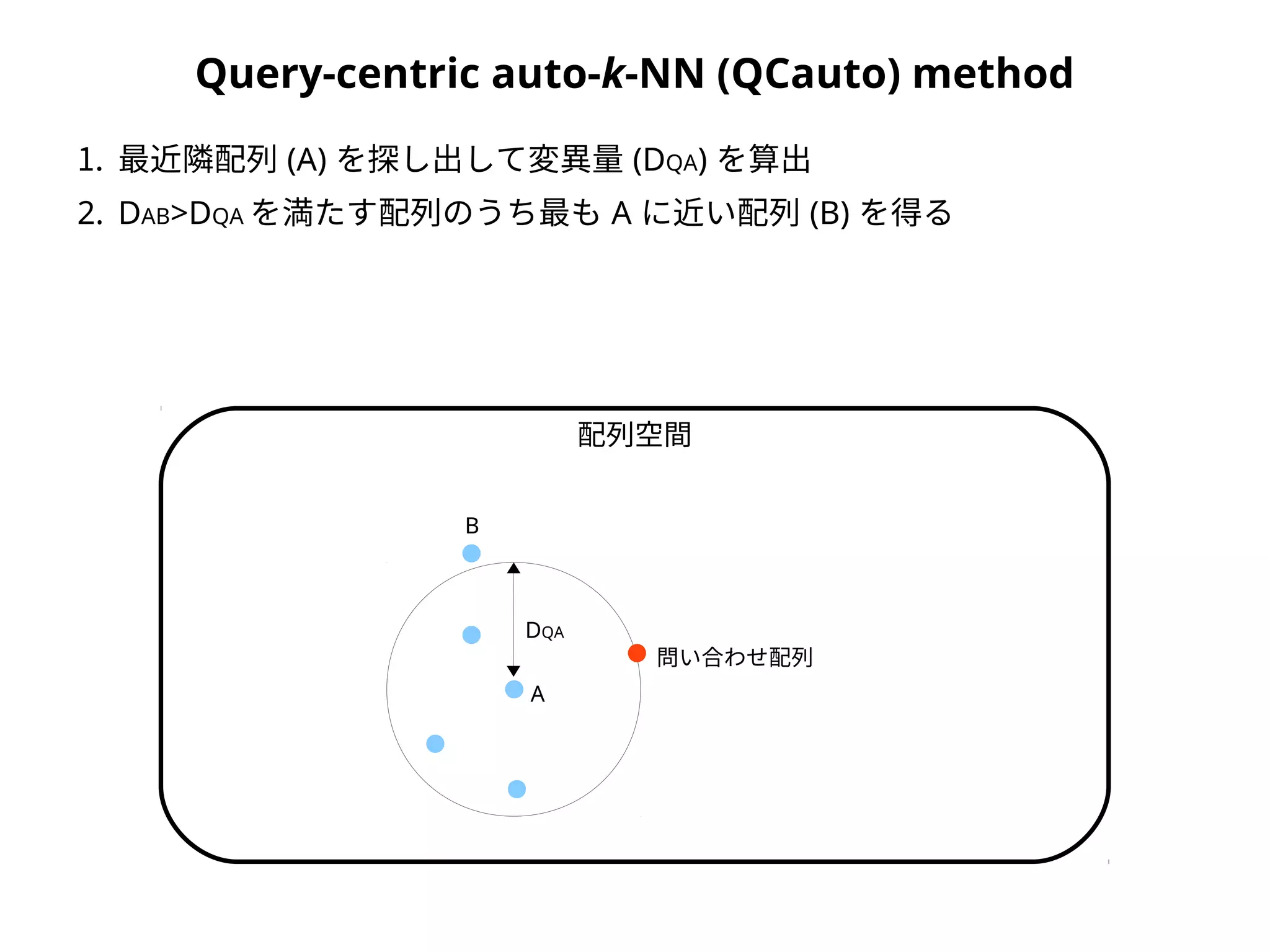
Query-centric auto-k-NN (QCauto)method
1. 最近隣配列 (A) を探し出して変異量 (DQA) を算出
2. DAB>DQA を満たす配列のうち最も A に近い配列 (B) を得る
A
DQA
問い合わせ配列
配列空間
B
144.
Query-centric auto-k-NN (QCauto)method
1. 最近隣配列 (A) を探し出して変異量 (DQA) を算出
2. DAB>DQA を満たす配列のうち最も A に近い配列 (B) を得る
3. DQN≤DQB を満たすすべての配列 (N) を得る
4. A, B, N の全配列で共通する分類群を採用
A
DQB
N
N
N
問い合わせ配列
配列空間
B
145.
Query-centric auto-k-NN (QCauto)method
1. 最近隣配列 (A) を探し出して変異量 (DQA) を算出
2. DAB>DQA を満たす配列のうち最も A に近い配列 (B) を得る
3. DQN≤DQB を満たすすべての配列 (N) を得る
4. A, B, N の全配列で共通する分類群を採用
A
DQB
N
N
N
問い合わせ配列
配列空間
B
問い合わせ配列と最近隣配列間の変異量
DQA
DQB
同定結果分類群内の最大変異量
<≤=































































































































































































![[DDBJing30] メタゲノム解析と微生物統合データベース](https://cdn.slidesharecdn.com/ss_thumbnails/30ddbjingmegap-141226013548-conversion-gate01-thumbnail.jpg?width=640&height=640&fit=bounds)






























































