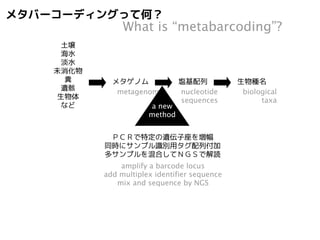
メタバーコーディングって何?
土壌
海水
淡水
未消化物
糞
遺骸
生物体
など
What is “metabarcoding”?
soils
sea water
fresh water
undigested materials
feces
dead bodies
living bodies
etc.
11.
メタバーコーディングって何?
土壌
海水
淡水
未消化物
糞
遺骸
生物体
など
What is “metabarcoding”?
メタゲノム
metagenomes
soils
sea water
fresh water
undigested materials
feces
dead bodies
living bodies
etc.
12.
メタバーコーディングって何?
土壌
海水
淡水
未消化物
糞
遺骸
生物体
など
What is “metabarcoding”?
メタゲノム塩基配列
metagenomes nucleotide
soils
sea water
fresh water
undigested materials
feces
dead bodies
living bodies
etc.
sequences
13.
メタバーコーディングって何?
土壌
海水
淡水
未消化物
糞
遺骸
生物体
など
What is “metabarcoding”?
メタゲノム塩基配列生物種名
metagenomes nucleotide
soils
sea water
fresh water
undigested materials
feces
dead bodies
living bodies
etc.
sequences
biological
taxa
14.
メタバーコーディングのポジティブスパイラル
A positivespiral of metabarcoding
メタバーコーディング
未知生物発見
従来法による記載分類
improve
metabarcoding
メタバーコーディング
能力向上
metabarcoding
DNA データベース充実
discover
a new taxon
describe the new taxon
by existing method
expand the DNA database
15.
メタバーコーディングって何?
土壌
海水
淡水
未消化物
糞
遺骸
生物体
など
What is “metabarcoding”?
メタゲノム塩基配列生物種名
metagenomes nucleotide
sequences
an old
method
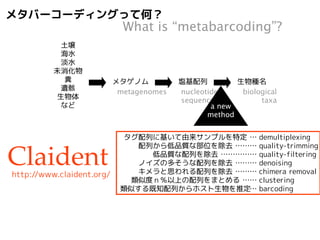
PCRで特定の遺伝子座を増幅
サンガー法で塩基配列解読
biological
taxa
amplify a barcode locus
sequence by Sanger method
16.
メタバーコーディングって何?
土壌
海水
淡水
未消化物
糞
遺骸
生物体
など
What is “metabarcoding”?
メタゲノム塩基配列生物種名
metagenomes nucleotide
sequences
a new
method
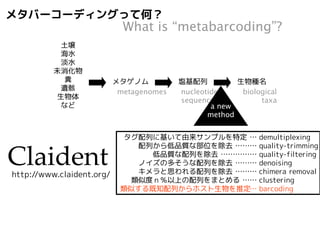
PCRで特定の遺伝子座を増幅
同時にサンプル識別用タグ配列付加
多サンプルを混合してNGSで解読
biological
taxa
amplify a barcode locus
add multiplex identifier sequence
mix and sequence by NGS
17.
メタバーコーディングって何?
土壌
海水
淡水
未消化物
糞
遺骸
生物体
など
What is “metabarcoding”?
メタゲノム塩基配列生物種名
metagenomes nucleotide
sequences
biological
an old
method
近縁既知配列と多重整列
分子系統樹推定
taxa
multiple alignment with known relatives
molecular phylogenetic inference
生物の所属分類群を特定=同定すると、様々なことがわかる
● 生態
Taxonomic identification gives us
a lot of information
Ecology
● 寿命,最大・平均・最小サイズ,生息環境,分布域, etc.
life history, body size, habitat, distribution, etc.
34.
生物の所属分類群を特定=同定すると、様々なことがわかる
● 生態
● 寿命,最大・平均・最小サイズ,生息環境,分布域, etc.
● 近縁の分類群
Taxonomic identification gives us
a lot of information
Ecology
life history, body size, habitat, distribution, etc.
Closely related taxa
35.
生物の所属分類群を特定=同定すると、様々なことがわかる
● 生態
Taxonomic identification gives us
a lot of information
Ecology
● 寿命,最大・平均・最小サイズ,生息環境,分布域, etc.
● 近縁の分類群
life history, body size, habitat, distribution, etc.
Closely related taxa
● ヒトであればチンパンジー,ライオンであればネコ, etc.
chimps for humans, lions for cats, etc.
より下位の階層まで同じ分類群は、より似ている
Organisms whichbelong same lower taxa
are more similar
● 既知生物に似ているほど下位の階層まで同定可能
An organism which is more similar to known organisms
is identifiable to lower taxa
38.
より下位の階層まで同じ分類群は、より似ている
Organisms whichbelong same lower taxa
are more similar
● 既知生物に似ているほど下位の階層まで同定可能
An organism which is more similar to known organisms
is identifiable to lower taxa
● より下位の階層(種とか)まで同定できるとより詳細にわかる
Lower taxonomic information provides
more detailed ecological information
新規準
A newcriterion for molecular identification
問い合わせ配列と最近隣配列間の変異量
distance between query and nearest-neighbor
44.
新規準
A newcriterion for molecular identification
問い合わせ配列と最近隣配列間の変異量
distance between query and nearest-neighbor
<
同定結果分類群内の最大変異量
maximum distance within resulting taxon
45.
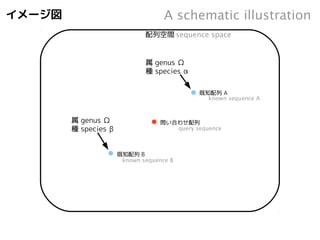
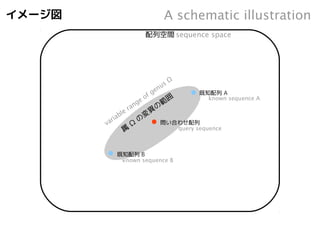
イメージ図
既知配列B
Aschematic illustration
sequence space
既知配列A
配列空間
問い合わせ配列
known sequence A
known sequence B
query sequence
46.
イメージ図
既知配列B
Aschematic illustration
既知配列A
属genus Ω
種species α
問い合わせ配列
属genus Ω
種species β
配列空間
sequence space
known sequence A
known sequence B
query sequence
47.
A schematic illustration
配列空間
属Ω の変異の範囲
known sequence B
イメージ図
既知配列B
sequence space
既知配列A
問い合わせ配列
known sequence A
query sequence
variable range of genus Ω
48.
A schematic illustration
known sequence A
属Ω の変異の範囲
known sequence B
query sequence
イメージ図
既知配列B
既知配列A
問い合わせ配列
属genus Ω
種species ?
配列空間
sequence space
variable range of genus Ω
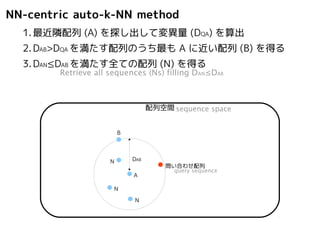
NN-centric auto-k-NN method
1.最近隣配列(A) を探し出して変異量(DQA) を算出
Retrieve nearest-neighbor (A), and calculate distance from query (DQA)
配列空間sequence space
問い合わせ配列
A
DQA
query sequence
51.
NN-centric auto-k-NN method
1.最近隣配列(A) を探し出して変異量(DQA) を算出
2.DAB>DQAを満たす配列のうち最もA に近い配列(B) を得る
Retrieve borderline (B),
which is nearest to A in the sequences farther from A than Q
DQA
A
配列空間
問い合わせ配列
B
sequence space
query sequence
52.
NN-centric auto-k-NN method
1.最近隣配列(A) を探し出して変異量(DQA) を算出
2.DAB>DQAを満たす配列のうち最もA に近い配列(B) を得る
3.DAN≤DABを満たす全ての配列(N) を得る
Retrieve all sequences (Ns) filling DAN≤DAB
DAB
A
B
N
N
N
配列空間sequence space
問い合わせ配列
query sequence
53.
NN-centric auto-k-NN method
1.最近隣配列(A) を探し出して変異量(DQA) を算出
2.DAB>DQAを満たす配列のうち最もA に近い配列(B) を得る
3.DAN≤DABを満たす全ての配列(N) を得る
4.A, B, N の全配列で共通する分類群を採用
Accept a taxon common to A, B, and Ns
DAB
A
B
N
N
N
配列空間sequence space
問い合わせ配列
query sequence
54.
NN-centric auto-k-NN method
1.最近隣配列(A) を探し出して変異量(DQA) を算出
distance between query and nearest-neighbor
2.DAB>DQAを満たす配列のうち問最いも合わA せ配に列近とい最近配隣列配列(B) 間のを変得異る
量
3.DAN≤DABを満たす全ての配列(N) を得る
4.A, B, N の全配列で共通する分類群を採用
< ≤ =
sequence space
DAB
A
B
N
N
N
配列空間
maximum distance within resulting taxon
問い合わせ配列
DQA
DAB
同定結果分類群内の最大変異量
query sequence
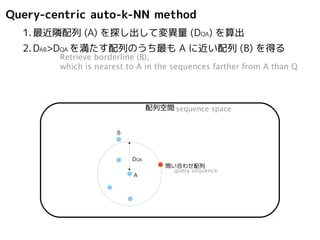
Query-centric auto-k-NN method
1.最近隣配列(A) を探し出して変異量(DQA) を算出
Retrieve nearest-neighbor (A), and calculate distance from query (DQA)
配列空間sequence space
問い合わせ配列
A
DQA
query sequence
57.
Query-centric auto-k-NN method
1.最近隣配列(A) を探し出して変異量(DQA) を算出
2.DAB>DQAを満たす配列のうち最もA に近い配列(B) を得る
Retrieve borderline (B),
which is nearest to A in the sequences farther from A than Q
DQA
A
配列空間
問い合わせ配列
B
sequence space
query sequence
58.
Query-centric auto-k-NN method
1.最近隣配列(A) を探し出して変異量(DQA) を算出
2.DAB>DQAを満たす配列のうち最もA に近い配列(B) を得る
3.DQN≤DQBを満たすすべての配列(N) を得る
Retrieve all sequences (Ns) filling DQN≤DQB
A
DQB
N
N
問い合わせ配列
N
配列空間
B
sequence space
query sequence
59.
Query-centric auto-k-NN method
1.最近隣配列(A) を探し出して変異量(DQA) を算出
2.DAB>DQAを満たす配列のうち最もA に近い配列(B) を得る
3.DQN≤DQBを満たすすべての配列(N) を得る
4.A, B, N の全配列で共通する分類群を採用
Accept a taxon common to A, B, and Ns
A
DQB
N
N
問い合わせ配列
N
配列空間
B
sequence space
query sequence
60.
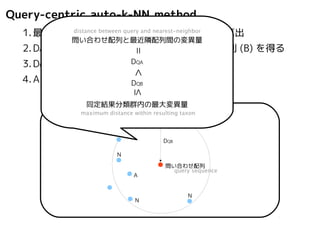
Query-centric auto-k-NN method
1.最近隣配列distance (A) between を探しquery 出しand てnearest-変異neighbor
量(DQA) を算出
問い合わせ配列と最近隣配列間の変異量
2.DAB>DQAを満たす配列のうち最もA に近い配列(B) を得る
3.DQN≤DQBを満たすすべての配列(N) を得る
4.A, B, N の全配列で共通する分類群を採用
sequence space
< ≤ =
maximum distance within resulting taxon
A
DQB
N
N
問い合わせ配列
N
配列空間
B
DQA
DQB
同定結果分類群内の最大変異量
query sequence
61.
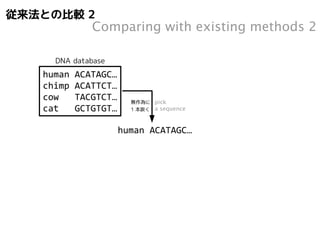
従来法との比較1
Comparing withexisting methods 1
DNA database
human ACATAGC…
chimp ACATTCT…
cow TACGTCT…
cat GCTGTGT…
62.
従来法との比較1
Comparing withexisting methods 1
DNA database
human ACATAGC…
chimp ACATTCT…
cow TACGTCT…
cat GCTGTGT… 無作為に
pick
1 本抜く
a sequence
human ACATAGC…
63.
従来法との比較1
Comparing withexisting methods 1
DNA database
無作為に
1 本抜く
DNA database lacking 1 sequence
human ACATAGC…
human ACATAGC…
chimp ACATTCT…
cow TACGTCT…
cat GCTGTGT…
chimp ACATTCT…
cow TACGTCT…
cat GCTGTGT…
pick
a sequence
64.
従来法との比較1
Comparing withexisting methods 1
DNA database
DNA database lacking 1 sequence
human ACATAGC…
human のDNA を
右のデータベースを使って
プログラムで同定して
正解かどうかを調べる
無作為に
1 本抜く
human ACATAGC…
chimp ACATTCT…
cow TACGTCT…
cat GCTGTGT…
chimp ACATTCT…
cow TACGTCT…
cat GCTGTGT…
pick
a sequence
identify human DNA by programs
with DNA database lacking human DNA
65.
従来法との比較1
Comparing withexisting methods 1
DNA database
DNA database lacking 1 sequence
human ACATAGC…
human のDNA を
右のデータベースを使って
プログラムで同定して
正解かどうかを調べる
無作為に
1 本抜く
human ACATAGC…
chimp ACATTCT…
cow TACGTCT…
cat GCTGTGT…
chimp ACATTCT…
cow TACGTCT…
cat GCTGTGT…
pick
a sequence
identify human DNA by programs
with DNA database lacking human DNA
抜き取ったDNA は
プログラムからは
未知のものになる
Picked DNA seems to be
“unknown sequence”
from programs
66.
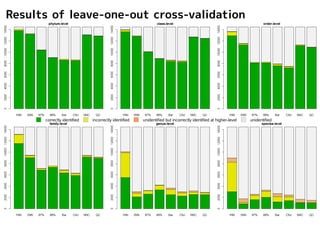
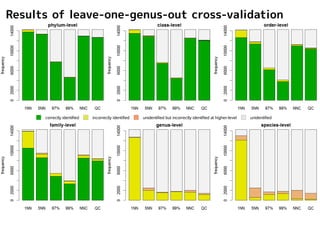
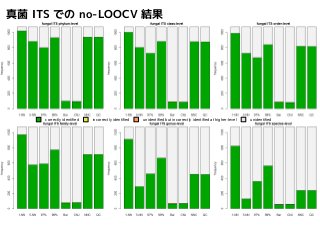
Results of leave-one-outcross-validation
correctly identified incorrectly identified unidentified but incorrectly identified at higher-level unidentified
67.
Results of leave-one-outcross-validation
1NN はよく落ちるが
誤同定多数
Too many misidentifications
were produced by 1NN method
correctly identified incorrectly identified unidentified but incorrectly identified at higher-level unidentified
68.
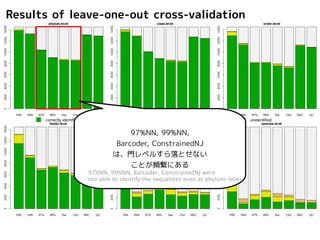
Results of leave-one-outcross-validation
correctly identified incorrectly identified unidentified but incorrectly identified at higher-level unidentified
97%NN, 99%NN,
Barcoder, ConstrainedNJ
は、門レベルすら落とせない
ことが頻繁にある
97%NN, 99%NN, Barcoder, ConstrainedNJ were
not able to identify the sequences even at phylum-level
69.
Results of leave-one-outcross-validation
correctly identified incorrectly identified unidentified but incorrectly identified at higher-level unidentified NNC, QC は門~科までは
よく落とせている
NNC and QC frequently produced correct identification
at phylum-, class-, order-, and family-level.
70.
Results of leave-one-outcross-validation
correctly identified incorrectly identified unidentified but incorrectly identified at higher-level unidentified
5NN はNNC, QC
とよく似た結果
5NN produced similar results to NNC and QC
71.
従来法との比較2
Comparing withexisting methods 2
DNA database
human ACATAGC…
chimp ACATTCT…
cow TACGTCT…
cat GCTGTGT… 無作為に
pick
1 本抜く
a sequence
human ACATAGC…
72.
従来法との比較2
Comparing withexisting methods 2
DNA database
無作為に
1 本抜く
DNA database lacking all seqs of a order
human ACATAGC…
human ACATAGC…
chimp ACATTCT…
cow TACGTCT…
cat GCTGTGT…
cow TACGTCT…
cat GCTGTGT…
pick
a sequence
73.
従来法との比較2
Comparing withexisting methods 2
DNA database
DNA database lacking all seqs of a order
human ACATAGC…
human のDNA を
右のデータベースを使って
プログラムで同定して
正解かどうかを調べる
無作為に
1 本抜く
human ACATAGC…
chimp ACATTCT…
cow TACGTCT…
cat GCTGTGT…
cow TACGTCT…
cat GCTGTGT…
pick
a sequence
identify human DNA by programs
with DNA database lacking all DNA of a order
74.
従来法との比較2
Comparing withexisting methods 2
DNA database
DNA database lacking all seqs of a order
human ACATAGC…
human のDNA を
右のデータベースを使って
プログラムで同定して
正解かどうかを調べる
無作為に
1 本抜く
human ACATAGC…
chimp ACATTCT…
cow TACGTCT…
cat GCTGTGT…
cow TACGTCT…
cat GCTGTGT…
pick
a sequence
identify human DNA by programs
with DNA database lacking all DNA of a order
抜き取ったDNA の
「目」は
プログラムからは
未知のものになる
Picked DNA seems to be
“unknown order”
from programs
75.
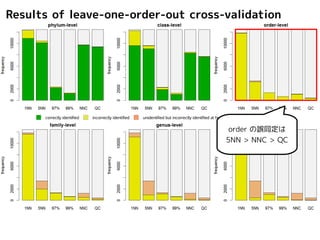
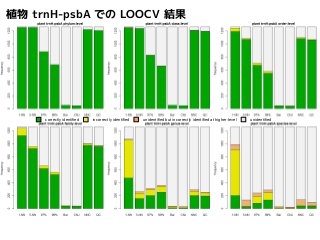
Results of leave-one-order-outcross-validation
correctly identified incorrectly identified unidentified but incorrectly identified at higher-level unidentified
76.
Results of leave-one-order-outcross-validation
correctly identified incorrectly identified unidentified but incorrectly identified at higher-l phylum, class ではevel unidentified
5NN, NNC, QC
はよく似た結果
77.
Results of leave-one-order-outcross-validation
correctly identified incorrectly identified unidentified but incorrectly identified at higher-level unidentified
order の誤同定は
5NN > NNC > QC
78.
Results of leave-one-order-outcross-validation
correctly identified incorrectly identified unidentified but incorrectly identified at higher-level unidentified
family でも
5NN > NNC > QC
79.
Results of leave-one-family-outcross-validation
correctly identified incorrectly identified unidentified but incorrectly identified at higher-level unidentified
80.
Results of leave-one-genus-outcross-validation
correctly identified incorrectly identified unidentified but incorrectly identified at higher-level unidentified
81.
まとめ
Conclusion
●QCauto method...
● 多重整列が不要
● 既知系統樹が不要
● どの遺伝子座でも適用可能
● どの分類群にも適用可能
● 前処理に時間がかからない
● 同定処理に時間がかからない
● 理論的背景がある
is multiple alignment free
is phylogenetic tree free
is locus independent
is taxon independent
is fast in preprocess
is fast in identification process
has theoretical background
● 「既知の綱の未知の目」といった答えを正しく出す
can find unknown order of existing class
82.
QCauto 法はポジティブスパイラルを加速する
QCautomethod accelerates positive spiral
メタバーコーディング
未知生物発見
従来法による記載分類
improve
metabarcoding
メタバーコーディング
能力向上
metabarcoding
DNA データベース充実
discover
a new taxon
describe the new taxon
by existing method
expand the DNA database
83.
QCauto 法はポジティブスパイラルを加速する
QCautomethod accelerates positive spiral
メタバーコーディング
未知生物発見
従来法による記載分類
improve
metabarcoding
メタバーコーディング
能力向上
metabarcoding
DNA データベース充実
discover
a new taxon
describe the new taxon
by existing method
expand the DNA database
Bottleneck























































































![[DDBJing30] メタゲノム解析と微生物統合データベース](https://cdn.slidesharecdn.com/ss_thumbnails/30ddbjingmegap-141226013548-conversion-gate01-thumbnail.jpg?width=640&height=640&fit=bounds)


















































![[2019-09-02] AI・IoT活用情報とGoogle Colab植物画像注釈](https://cdn.slidesharecdn.com/ss_thumbnails/plantinformatics190902-190906044014-thumbnail.jpg?width=640&height=640&fit=bounds)








![[2021-03-14] 植物表現型画像解析のための手作業注釈加速化手法とActive Learning](https://cdn.slidesharecdn.com/ss_thumbnails/ek210314vfin-210804060823-thumbnail.jpg?width=640&height=640&fit=bounds)

![[DDBJing29]DDBJ, NIG SuperComputer, 大量配列情報解析(第29回 DDBJing 講習会 in 三島)](https://cdn.slidesharecdn.com/ss_thumbnails/29ddbjjingyn-140624004739-phpapp02-thumbnail.jpg?width=640&height=640&fit=bounds)


![[DDBJing30] DDBJ と NIG SuperComputer の紹介、大量配列情報](https://cdn.slidesharecdn.com/ss_thumbnails/30ddbjingddbj-141224194318-conversion-gate02-thumbnail.jpg?width=640&height=640&fit=bounds)








