






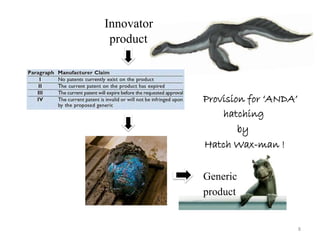
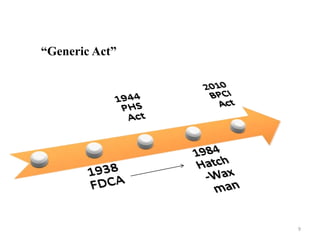





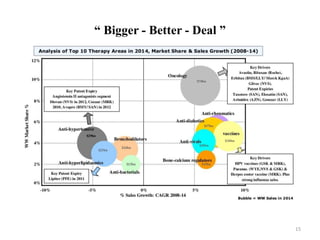
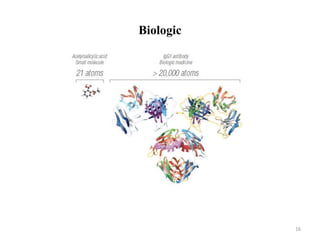

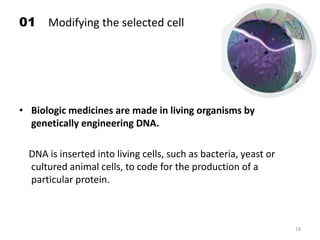



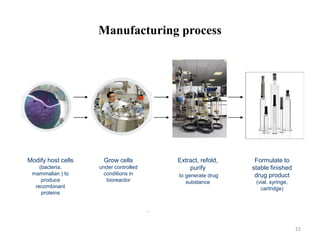




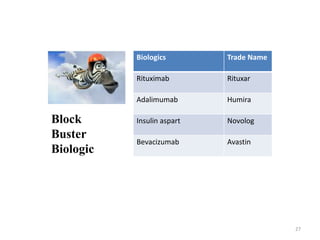




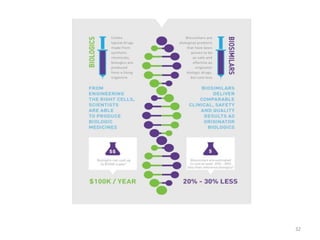
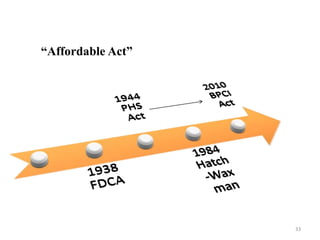

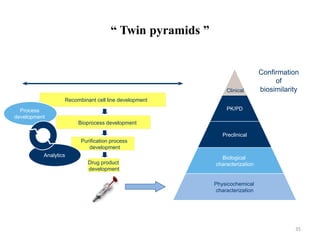

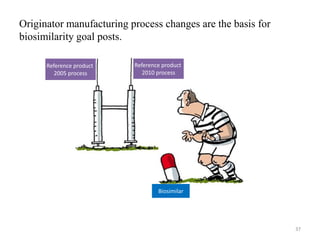
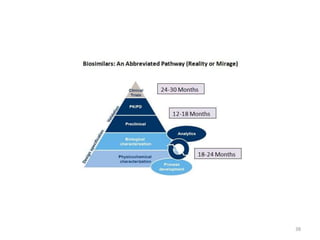




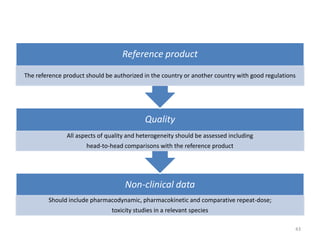






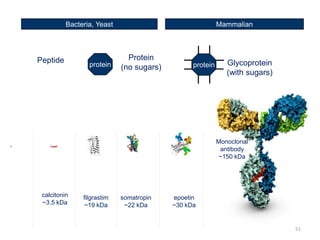



The document discusses generics and biosimilars. It provides background on the Hatch-Waxman Act which established the abbreviated new drug application process for generic small molecule drugs. It also discusses the Biologics Price Competition and Innovation Act which created an abbreviated licensure pathway for biosimilar biologics. The manufacturing of biologics is more complex than small molecules due to production in living cells, and biosimilars are highly similar but not identical copies. Clinical trials are required to demonstrate biosimilarity in terms of safety and efficacy.







































![Bio pharmaceutical classification System [BCS]](https://cdn.slidesharecdn.com/ss_thumbnails/biopharmaceuticalclassificationystembcs-160328061345-thumbnail.jpg?width=640&height=640&fit=bounds)















































