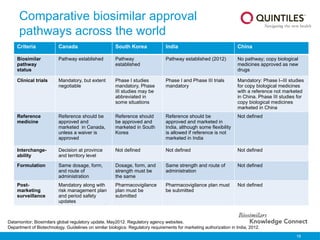
The document presents an overview of biosimilars, highlighting their definition, development, and impact on healthcare costs and access to treatments. It notes that biosimilars are similar to originator biological medicines but undergo a more rigorous approval process, reflecting their complex nature and manufacturing challenges. The document also discusses the increasing approval rates of biosimilars in Europe and their potential to lower drug costs significantly, providing examples of approved products and their market effects.








![13
http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_000408.jsp&mid=WC0b01ac058002958c
The BPCI Act appears in Title VII, subtitle A of the Patient Protection and Affordable Care Act, March 2010.
US Food and Drug Administration. Guidance for industry. Scientific considerations in demonstrating biosimilarity to a reference
product. Draft Guidance. Feb 2012.
Draft revisions
to Overarching
Guideline;
Quality
Guideline;
Non-clinical
and Clinical
Guideline
2004 2009 2011
2006 2007 2008 2012
2010 2013
2005
EMEA Legislative
Pathway
EMEA Regulatory
Guidance [Overarching
Guideline] under
revision
Product Class
Specific Guidelines:
Low molecular
weight heparin,
recombinant
Interferon-alpha
Product Class
Monoclonal
antibodies –
non-clinical and
clinical issues
Product Class
Specific Guideline:
Erythropoietin
(revised)
Quality Guideline;
Non-Clinical and
Clinical Guideline
(under revision )
Product Class
Specific Guidelines:
Insulin, G-CSF,
Somatropin
Product Class
Immunogenicity
assessment of
monoclonal
antibodies
Public Health
Service Act
amended to
allow the
approval of
biosimilars
Overarching Draft
Guidelines on
biosimilars
Europe US
Biosimilar regulations in EU and
USA: different stages of development
• The EU pioneered the development of
biosimilar regulations
• US overarching guidelines issued
Draft revisions to
Product Class
Specific Guideline:
Insulin, low molecular
weight heparin
Product
Class
Follicle
stimulating
hormone,
Interferon-
beta](https://image.slidesharecdn.com/biosimilarsknowledgeconnectslideresource-241215082617-9954531e/85/Biosimilars-Knowledge-Connect-slide-resource-pptx-13-320.jpg)