Downloaded 12 times




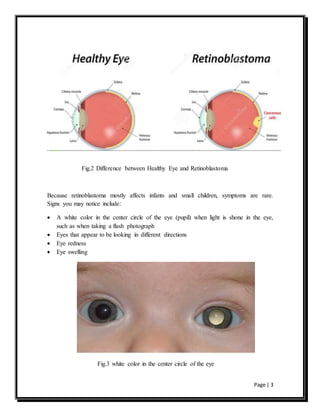




Retinoblastoma is a rare eye cancer primarily affecting young children, caused by mutations in the rb1 gene which disrupt normal cell growth in the retina. It can be hereditary, increasing the risk of tumors in both eyes and other cancers, or sporadic, typically resulting in a single tumor in one eye. Understanding the genetic basis of retinoblastoma enhances awareness of its inheritance patterns and the potential need for monitoring for additional cancers in affected individuals.






























































































![CTEV [ clubfoot] DR ARUN LAL ,DR MOHAMED ASHRAF travancore medical college k...](https://cdn.slidesharecdn.com/ss_thumbnails/ctevclubfootdrarunlaldrmohamedashraftravancoremedicalcollegekollamkeralaindia-260208063247-18fc466c-thumbnail.jpg?width=640&height=640&fit=bounds)












