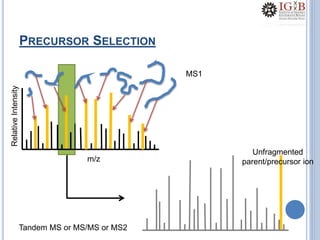
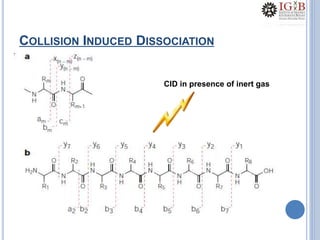
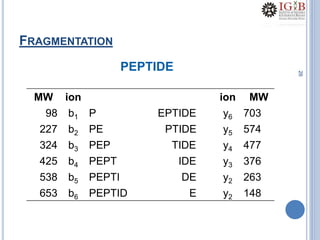
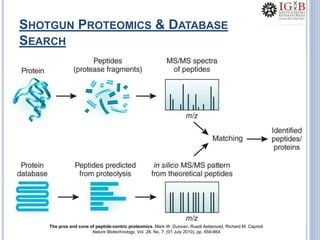
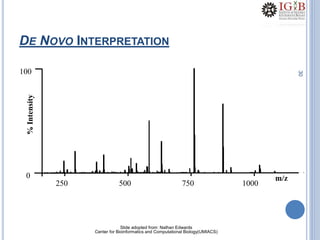
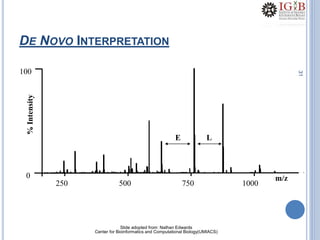
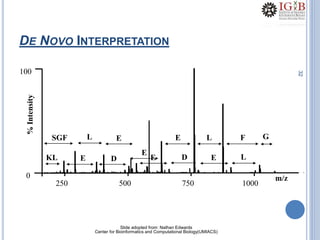
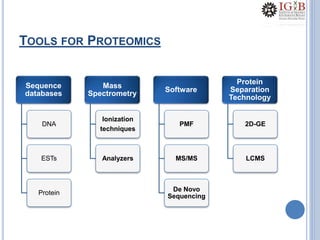
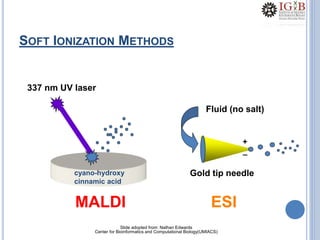
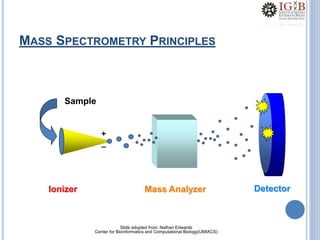
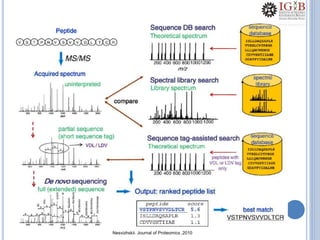
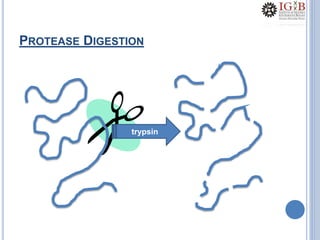
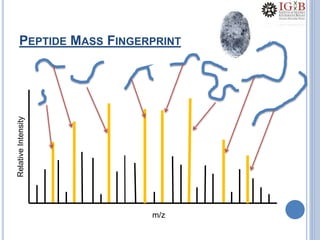
Proteomics uses mass spectrometry to characterize proteins in a biological sample on a large scale. It provides information about which proteins are present, how much of each protein exists, and how proteins are modified. Mass spectrometry coupled with protein separation techniques and database searching represents a major advancement for protein analysis, allowing characterization of entire proteomes. Key aspects of proteomics include separating proteins, ionizing peptides using techniques like MALDI and ESI, and identifying proteins by matching mass spectra to in silico digests or sequencing peptides de novo. While sample preparation and computational methods are still developing, proteomics offers insights beyond what is possible with genomics alone.


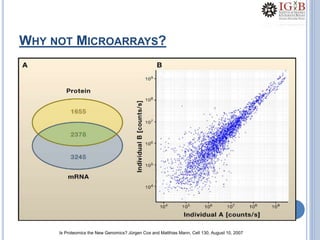




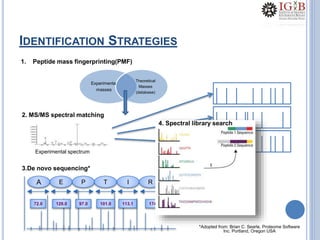


![PMF DATABASE SEARCH
450.2201
609.3667
698.3100
1007.5391
1199.4916
2098.9909
PEAKLIST
>gi|2924450|emb|CAA17750.1| PROBABLE FATTY-ACID-CoA LIGASE FADD18 (FRAGMENT) (FATTY-ACID-CoA
SYNTHETASE) (FATTY-ACID-CoA SYNTHASE) [Mycobacterium tuberculosis H37Rv]
MAASLSENLSCHSSNMCRLSGNAATNLERPGEEPPGDRCTRRQAVRPARTLAKKGNIPVGYYKDEKKTAETFRTINGVRYAIPGD
YAQVEEDGTVTMLGRGSVSINSGGEKVYPEEVEAALKGHPDVFDALVVGVPDPRY
GQQVAAVVQARPGCRPSLAELDSFVRSEIAGYKVPRSLWFVDEVKRSPAGKPDYRWAKEQTEARPADDVH
AGHVTSGS
>gi|15610649|ref|NP_218030.1| fatty-acid-CoA ligase [Mycobacterium tuberculosis H37Rv]
MAASLSENLSCHSSNMCRLSGNAATNLERPGEEPPGDRCTRRQAVRPARTLAKKGNIPVGYYKDEKKTAE
TFRTINGVRYAIPGDYAQVEEDGTVTMLGRGSVSINSGGEKVYPEEVEAALKGHPDVFDALVVGVPDPRY
GQQVAAVVQARPGCRPSLAELDSFVRSEIAGYKVPRSLWFVDEVKRSPAGKPDYRWAKEQTEARPADDVH
AGHVTSGS
Protein FASTA
database
450.2017 (P21234)
609.2667 (P12345)
664.3300 (P89212)
1007.4251 (P12345)
1114.4416 (P89212)
1183.5266 (P12345)
1300.5116 (P21234)
1407.6462 (P21234)
1526.6211 (P89212)
1593.7101 (P89212)
1740.7501 (P21234)
2098.8909 (P12345)
in silico
digestion
OUTPUT:
2 Unknown masses
1 hit on P21234
3 hits on P12345
RESULT:
protein is P12345](https://image.slidesharecdn.com/1-141219000811-conversion-gate02/85/1-proteomics-coursework-3-dec2012-aky-21-320.jpg)