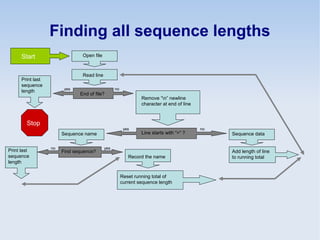
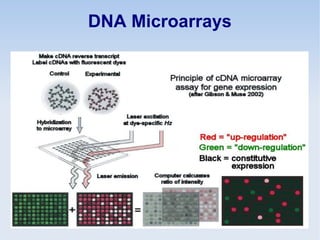
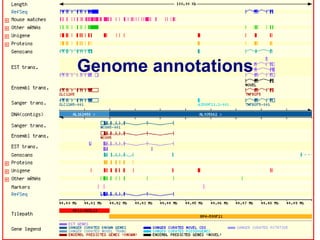
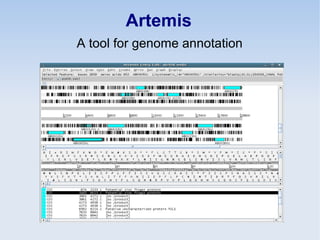
This document discusses the role of programming in computational biology. It begins by describing different types of programming languages like imperative, object-oriented, and functional languages. It then discusses how programming can reduce time, money, effort and errors in computational biology applications. Some key applications of programming in computational biology mentioned are data mining, genome annotation, microarray analysis, phylogenetics, and next generation sequencing studies. The document also discusses popular bioinformatics programming languages like Perl and describes concepts in programming like objects, modules, and the common gateway interface.















![Normalizing by column
Remaps gene arrays to column arrays
($experiment, $expr)
= read_expr ("expr.txt");
my @genes = sort keys %$expr;
for ($i = 0; $i < @$experiment; ++$i) {
my @col;
foreach $j (0..@genes-1) {
$col[$j] = $expr->{$genes[$j]}->[$i];
}
normalize_array(@col);
foreach $j (0..@genes-1) {
$expr->{$genes[$j]}->[$i] = $col[$j];
}
}
Puts column
data in @col
Puts @col
back into %expr
Normalizes (note use
of reference)](https://image.slidesharecdn.com/seminar-12877631631309-phpapp02/85/Programming-in-Computational-Biology-20-320.jpg)




![Self-intersection of a GFF file
sub self_intersect_GFF {
my @gff = @_;
my @intersect;
foreach $igff (@gff) {
foreach $jgff (@gff) {
if ($igff ne $jgff) {
if ($$igff[0] eq $$jgff[0]) {
if (!($$igff[3] > $$jgff[4]
|| $$jgff[3] > $$igff[4])) {
push @intersect, $igff;
last;
}
}
}
}
}
return @intersect;
}
Note: this code is slow.
Vast improvements in
speed can be gained if
we sort the @gff array
before checking for
intersection.
Fields 0, 3 and 4 of the
GFF line are the sequence
name, start and end co-
ordinates of the feature](https://image.slidesharecdn.com/seminar-12877631631309-phpapp02/85/Programming-in-Computational-Biology-27-320.jpg)
![Converting GFF to sequence
• Puts together several previously-described subroutines
• Namely: read_FASTA read_GFF revcomp print_seq
($gffFile, $seqFile) = @ARGV;
@gff = read_GFF ($gffFile);
%seq = read_FASTA ($seqFile);
foreach $gffLine (@gff) {
$seqName = $gffLine->[0];
$seqStart = $gffLine->[3];
$seqEnd = $gffLine->[4];
$seqStrand = $gffLine->[6];
$seqLen = $seqEnd + 1 - $seqStart;
$subseq = substr ($seq{$seqName}, $seqStart-1, $seqLen);
if ($seqStrand eq "-") { $subseq = revcomp ($subseq); }
print_seq ("$seqName/$seqStart-$seqEnd/$seqStrand", $subseq);
}](https://image.slidesharecdn.com/seminar-12877631631309-phpapp02/85/Programming-in-Computational-Biology-28-320.jpg)





![OOP: Jargon
• Member, method
– A variable/subroutine associated with a particular class
• Overriding
– When a derived class implements a method differently from its parent
class
• Constructor, destructor
– Methods called when an object is created/destroyed
• Accessor
– A method that provides [partial] access to hidden data
• Factory
– An [abstract] object that creates other objects
• Singleton
– A class which is only ever instantiated once (i.e. there’s only ever one
object of this class)
– C.f. static member variables, which occur once per class](https://image.slidesharecdn.com/seminar-12877631631309-phpapp02/85/Programming-in-Computational-Biology-36-320.jpg)