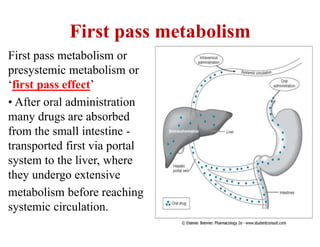
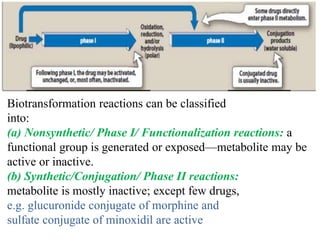
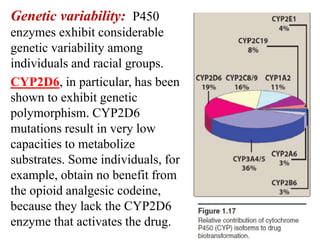
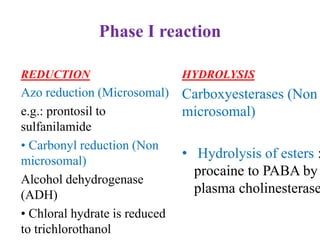
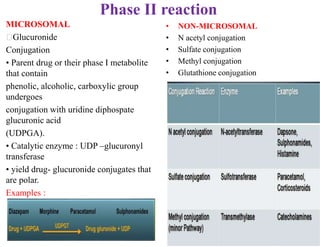
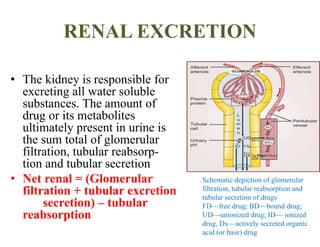
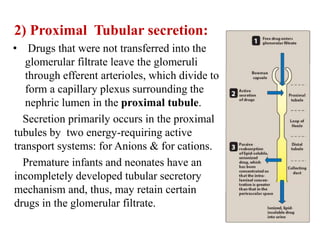
The document discusses pharmacokinetics, focusing on the biotransformation and elimination of drugs in the body, emphasizing the role of the liver in drug metabolism. It covers the significance of prodrugs, first-pass metabolism, and distinguishes between phase I and phase II reactions, detailing the involvement of cytochrome P450 enzymes. Additionally, it addresses factors affecting drug metabolism and the importance of renal excretion, highlighting how drug characteristics and biological factors influence pharmacokinetic processes.