Downloaded 40 times




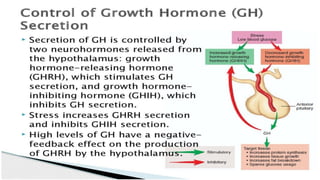



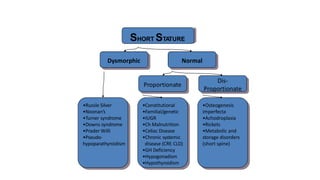



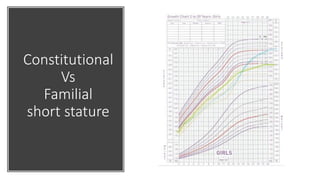





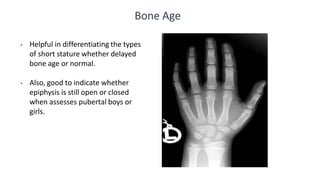





























This document provides information on pediatric growth hormone deficiency. It discusses GH secretion and control, GH physiology, the metabolic effects of GH, and approaches to evaluating and diagnosing a child with short stature. Causes of GH deficiency include defects in the hypothalamus or pituitary gland due to genetic mutations, injuries, or tumors. GH stimulation tests are used to diagnose GH deficiency. Treatment involves recombinant human GH therapy to improve growth and development.























































































