Downloaded 48 times



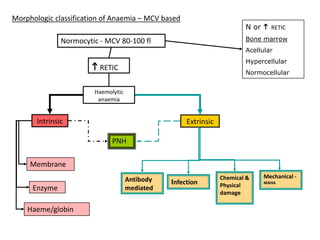
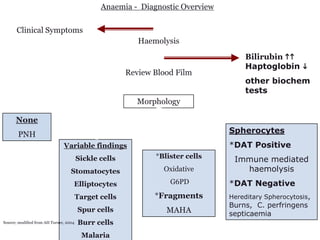





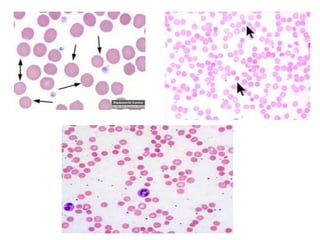

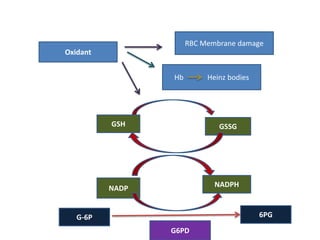



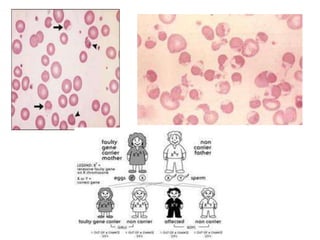




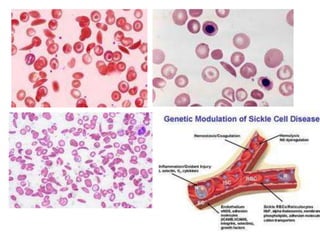


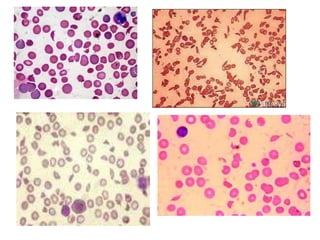

This document provides an overview of different types of anaemia, including classifications based on red blood cell size (MCV) and the underlying causes and mechanisms of haemolytic anaemias. Specific examples discussed include hereditary spherocytosis, G6PD deficiency, sickle cell disease, and microangiopathic haemolytic anaemia. Diagnostic findings and key features are outlined for each condition.




















































































