SYSTEMIC COMPLICATIONS OF
ORTHOPAEDICS
:SHOCK,CRUSH SYNDROME,DIC,ARDS
DR BIPUL BORTHAKUR
PROFFESOR AND HEAD
DEPARTMENT OF ORTHOPAEDICS ASSAM MEDICAL COLLEGE
DIBRUGARH , ASSAM
INTRODUCTION
• Trauma -majorcause of death and disability worldwide
• Deaths d/t trauma - trimodal pattern,
• 50% of fatally injured casualties died from non-survivable injuries
immediately
• 30% survived the initial trauma, but died within 1–3 hours;
• the remaining 20% died -complications at a late stage during the
6 weeks after injury
4.
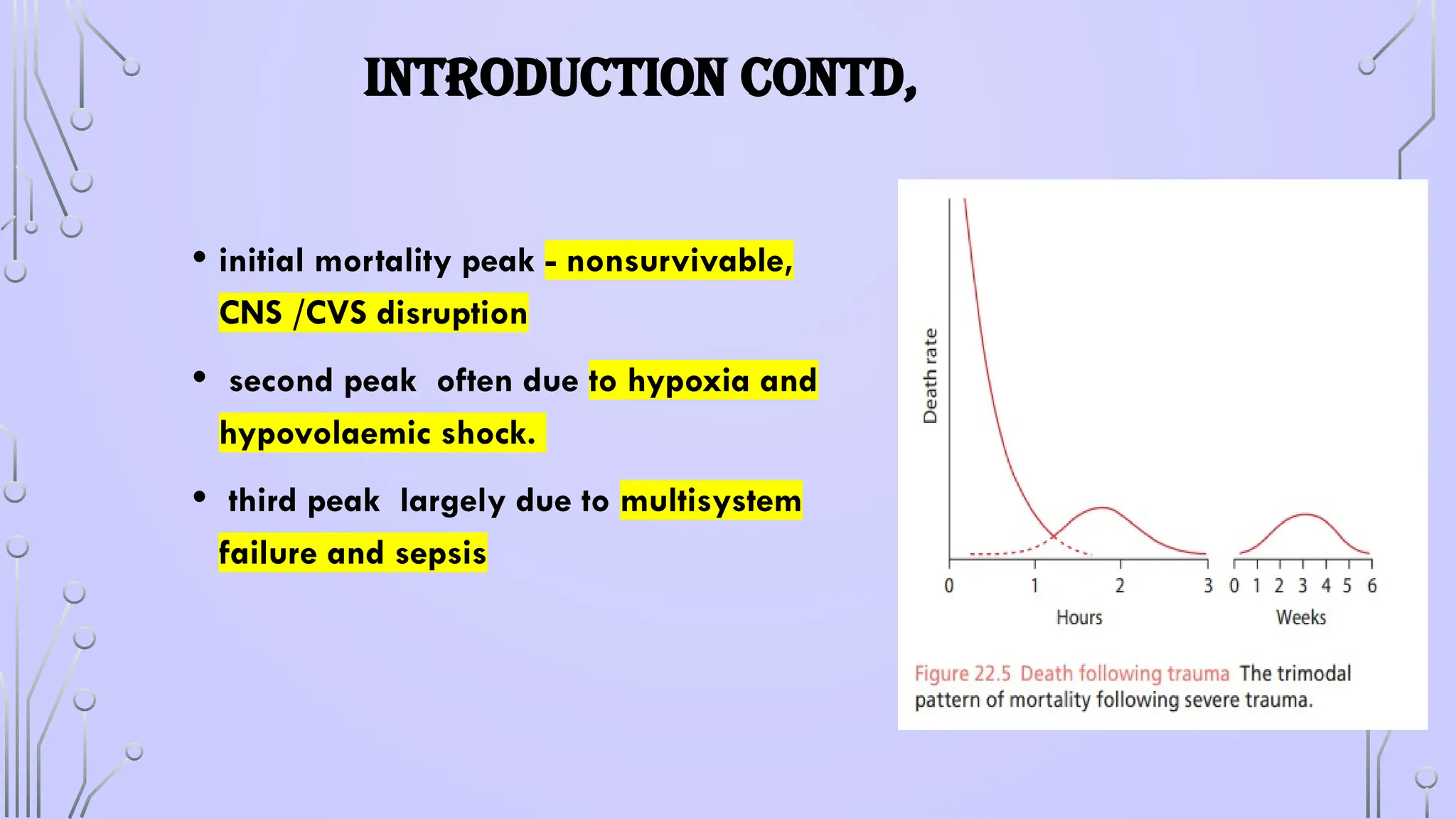
INTRODUCTION CONTD,
• initialmortality peak - nonsurvivable,
CNS /CVS disruption
• second peak often due to hypoxia and
hypovolaemic shock.
• third peak largely due to multisystem
failure and sepsis
5.
PATHOPHYSIOLOGY AND IMMUNERESPONSE TO TRAUMA
•physiologic response to injury 3 phases:
• (a) hypodynamic ebb phase (shock) ---- body initially attempts to limit the
blood loss to maintain perfusion to the vital organs;
• (b) a hyperdynamic flow ( up to 2 weeks) blood flow, in order to remove
waste products & allow nutrients to reach the site of injury for repair
• (c) recuperation phase (for months) to allow the human body to attempt to
return to its pre-injury level.
6.
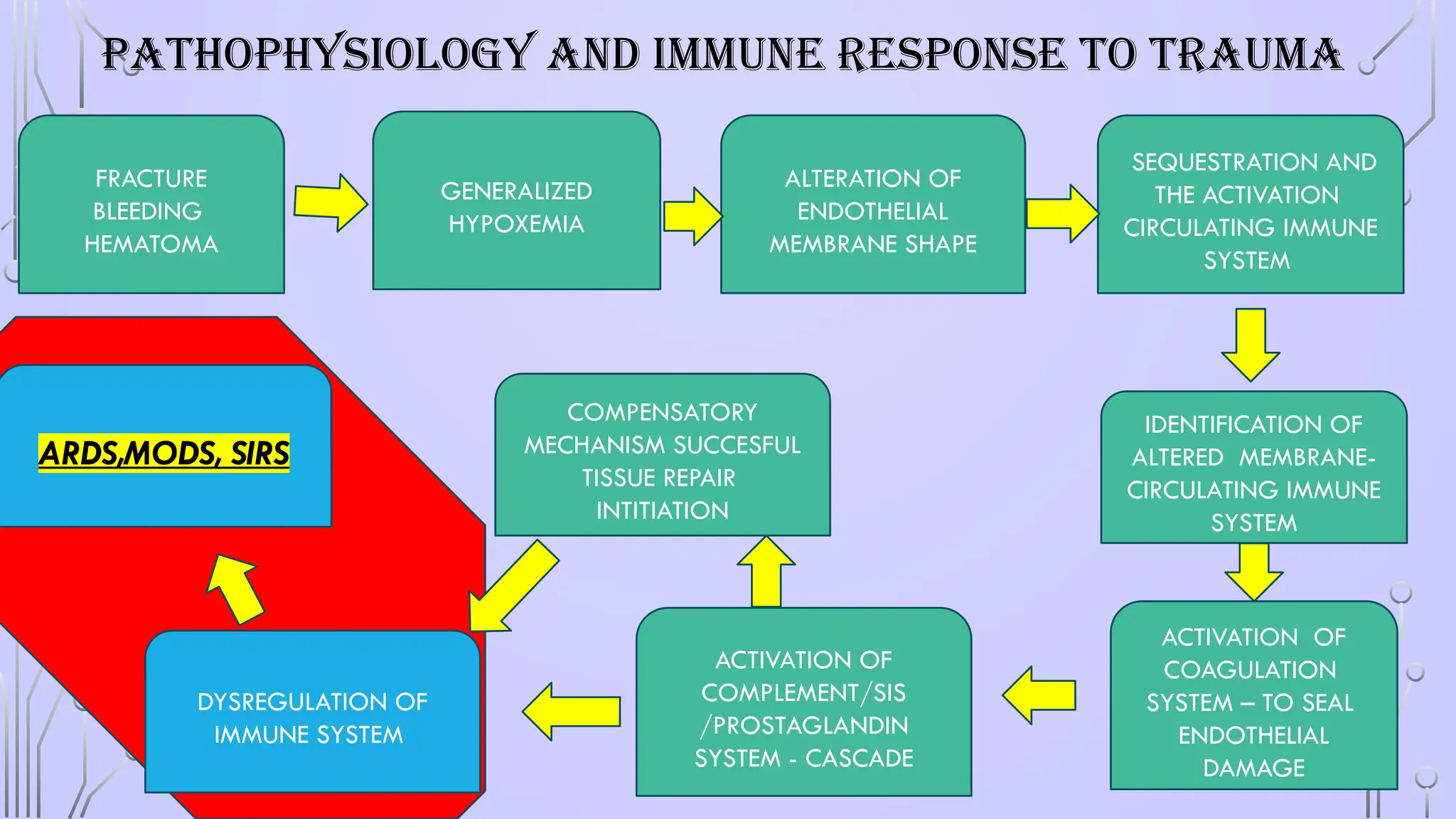
PATHOPHYSIOLOGY AND IMMUNERESPONSE TO TRAUMA
FRACTURE
BLEEDING
HEMATOMA
GENERALIZED
HYPOXEMIA
ALTERATION OF
ENDOTHELIAL
MEMBRANE SHAPE
IDENTIFICATION OF
ALTERED MEMBRANE-
CIRCULATING IMMUNE
SYSTEM
SEQUESTRATION AND
THE ACTIVATION
CIRCULATING IMMUNE
SYSTEM
ARDS,MODS, SIRS
ACTIVATION OF
COMPLEMENT/SIS
/PROSTAGLANDIN
SYSTEM - CASCADE
ACTIVATION OF
COAGULATION
SYSTEM – TO SEAL
ENDOTHELIAL
DAMAGE
DYSREGULATION OF
IMMUNE SYSTEM
COMPENSATORY
MECHANISM SUCCESFUL
TISSUE REPAIR
INTITIATION
8.
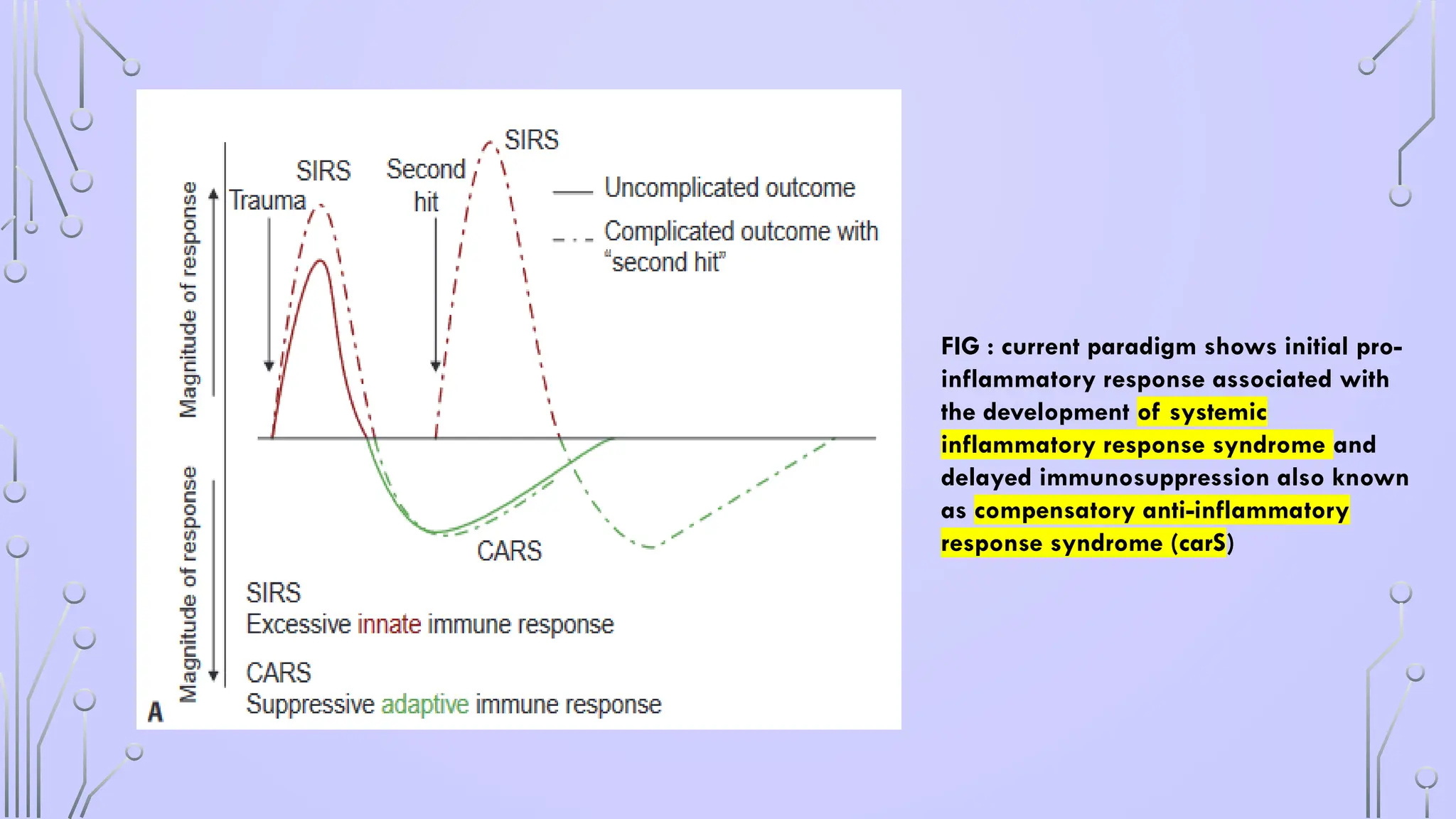
FIG : currentparadigm shows initial pro-
inflammatory response associated with
the development of systemic
inflammatory response syndrome and
delayed immunosuppression also known
as compensatory anti-inflammatory
response syndrome (carS)
9.
SHOCK
• state ofdecreased perfusion of the body resulting in inadequate
supply of oxygen and nutrients to the tissues leading to dysfunction
of the normal cellular physiology
• Requires Immediate treatment
• inadequate tissue oxygenation leads to irreversible cell injury and
death.
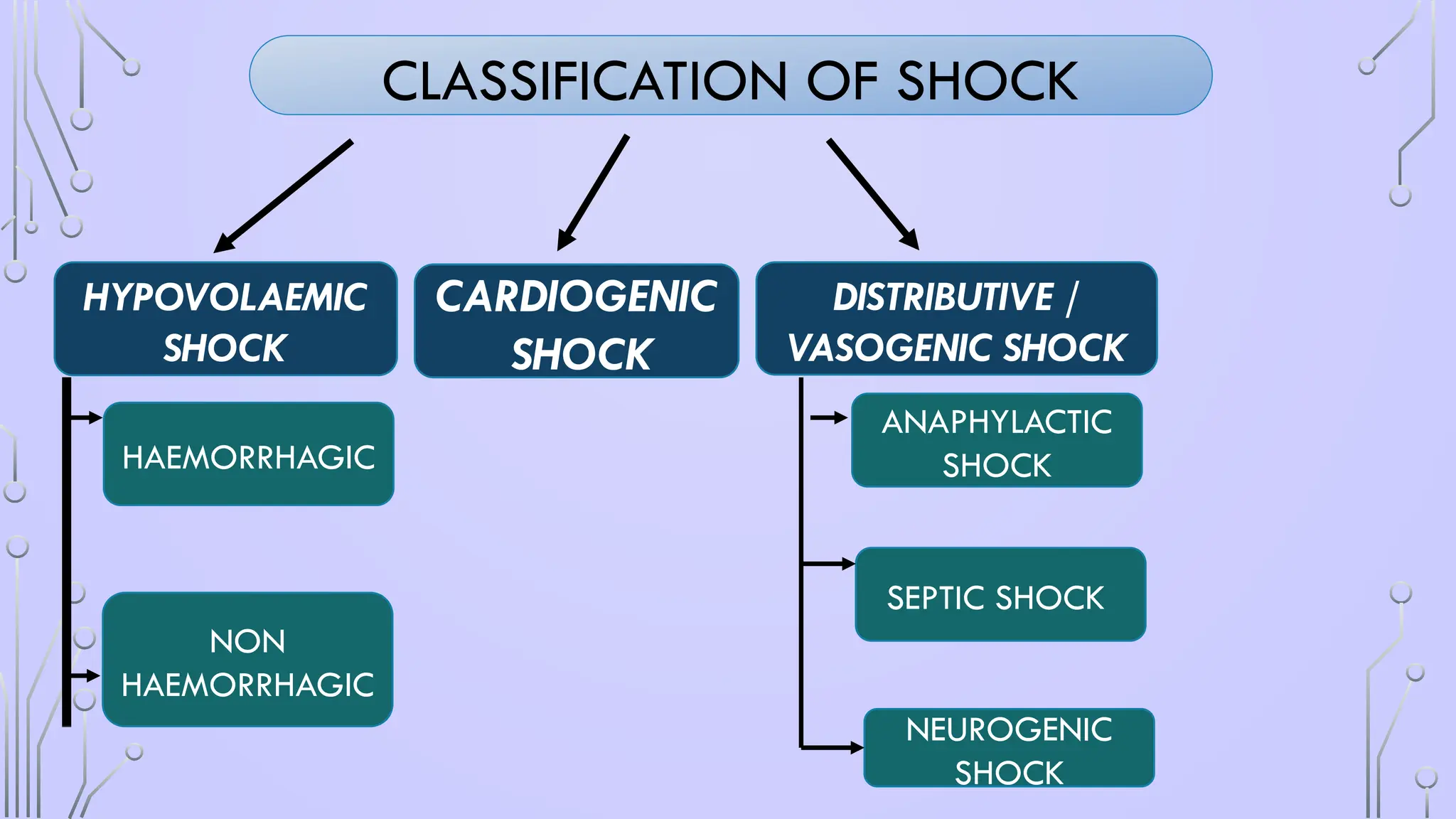
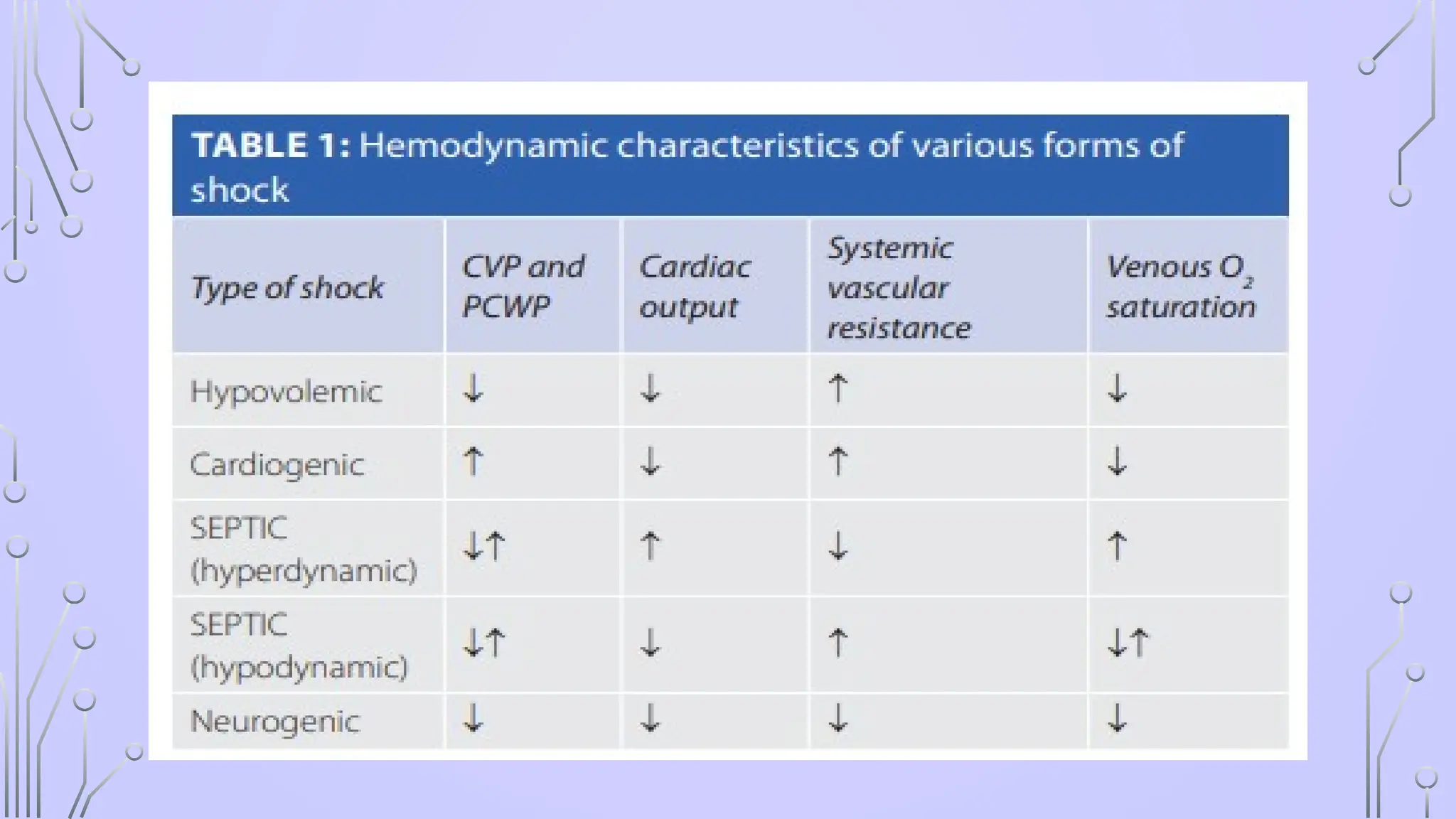
HYPOVOLAEMIC SHOCK
• Decreasedintravascular volume resulting from loss of blood,
plasma or fluids and electrolytes
• hypotension occurs, peripheral resistance increases, capillary
and venous beds collapse, and the tissue progressively
becomes hypoxic.
13.
HYPOVOLAEMIC SHOCK
• 2TYPES
• HEMORRHAGIC HYPOVOLEMIA :
• m/c cause of shock in trauma ,
• decreased cardiac output leading to hypoperfusion of the tissues and cell
death.
• NONHEMORRHAGIC HYPOVOLEMIC SHOCK
• massive gastrointestinal (GI) fluid or urinary losses leading to severe
dehydration.
• diversion of fluid into the extravascular compartments, commonly referred to
as “third spacing”
• systemic inflammation, acute pancreatitis, hepatic failure, surgery, burns or
intestinal obstruction.
15.
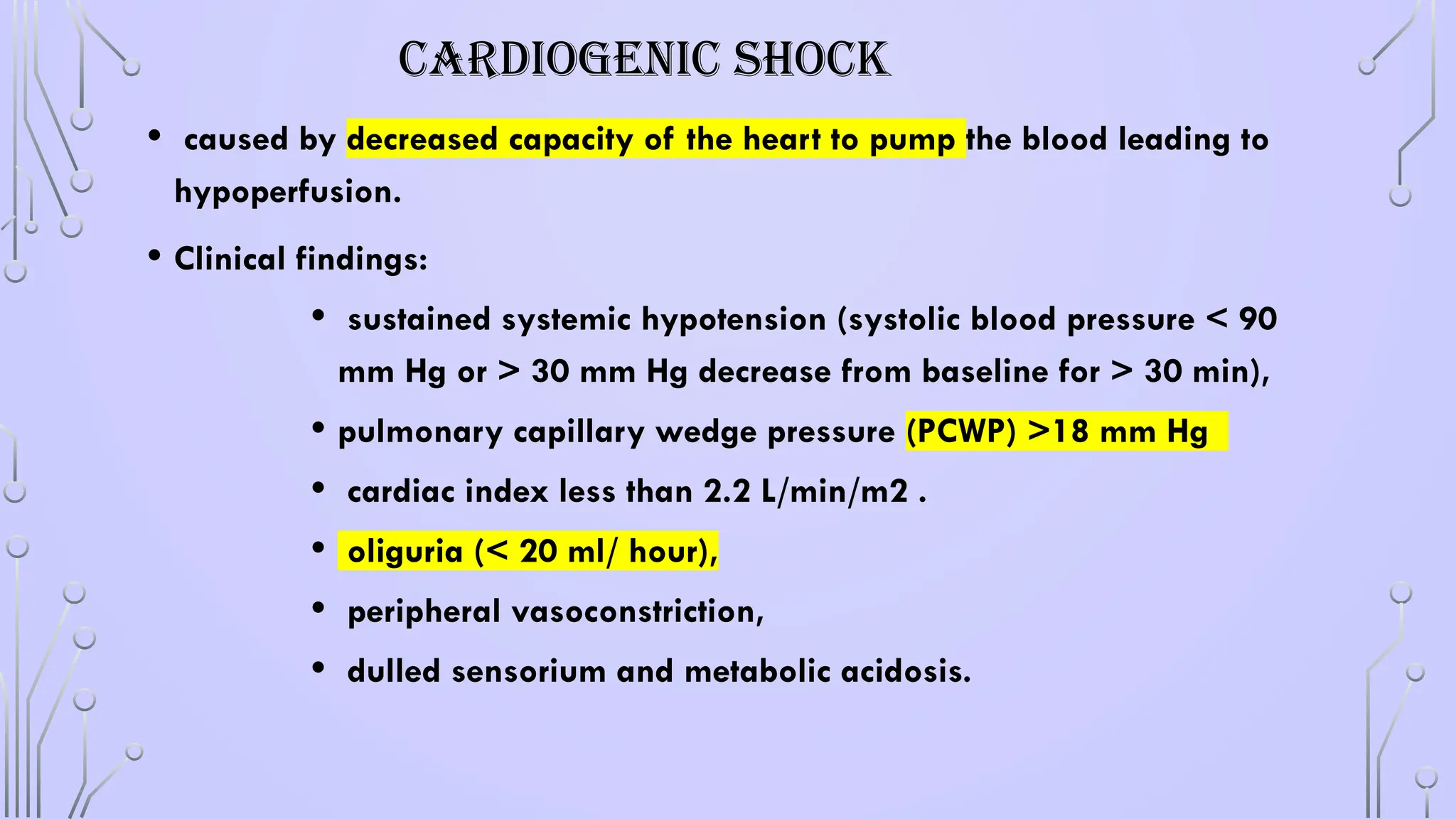
CARDIOGENIC SHOCK
• causedby decreased capacity of the heart to pump the blood leading to
hypoperfusion.
• Clinical findings:
• sustained systemic hypotension (systolic blood pressure < 90
mm Hg or > 30 mm Hg decrease from baseline for > 30 min),
• pulmonary capillary wedge pressure (PCWP) >18 mm Hg
• cardiac index less than 2.2 L/min/m2 .
• oliguria (< 20 ml/ hour),
• peripheral vasoconstriction,
• dulled sensorium and metabolic acidosis.
16.
CAUSES OF CARDIOGENICSHOCK
• acute myocardial infarction/ischemia
• blunt trauma to the heart (tension pneumothorax, cardiac
tamponade)
• tachyarrhythmias
• acute fulminant myocarditis
• HCM with severe outflow obstruction
• pulmonary embolus
• severe valvular heart
• Aortic dissection with aortic insufficiency or tamponade
• beta-blocker or calcium channel antagonist overdose.
17.
DISTRIBUTIVE SHOCK
• Reductionin systemic vascular resistance from diverse etiologies result in
inadequate cardiac output despite normal circulatory volume.
• There are three types:
(i) septic shock
(ii) neurogenic shock
(III) Anaphylactic shock.
18.
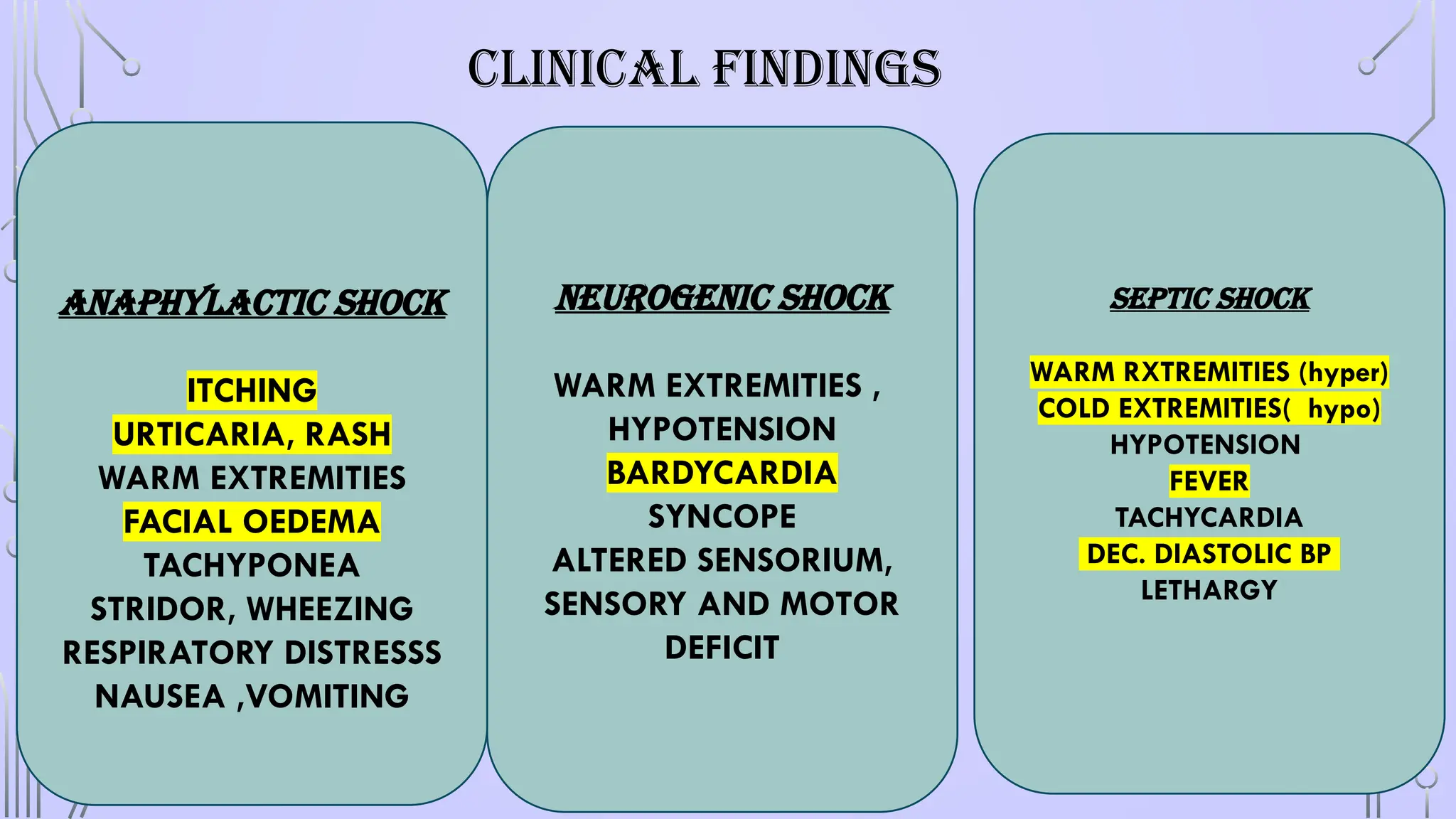
SEPTIC SHOCK
• “Septicshock” is defined as severe sepsis with hypotension in spite of fluid
resuscitation.
• Sepsis can be a response to any class of microorganism..
• the proinflammatory state, Release of mediators like cytokines. Complement,
oxygen radicals etc. leading to profound vasodilation, capillary leak and loss
of intravascular volume.
• Commonly gram-negative organisms like Escherichia coli, Klebsiella, Proteus, and
Pseudomonas
• incidence of sepsis from Gram-positive bacteria (e.g. S. aureus) and fungal
organisms is increasing.
19.
ANAPHYLACTIC SHOCK
• Alife-threatening allergic reaction that causes shock (hypoperfusion) and airway swelling.
• “Anaphylactic shock” is a term that specifically refers to an episode of anaphylaxis.
MECHANISM OF ANAPHYLACTIC SHOCK
1) NON IMMUNULOGICAL
2) IMMUNOLOGICAL
IgE mediated – histamine release
Non IgE mediated -
20.
NEUROGENIC SHOCK
• Interruptionof sympathetic vasomotor input results in loss of sympathetic tone with a
reduction in systemic vascular resistance and hypotension without a compensatory
tachycardia
• hypotension, bradycardia and syncope
• causes
• following injury to cervical or upper thoracic spine
• inadvertent cephalad migration of spinal anesthesia
• devastating head injury - interruption of sympathetic vasomotor input.
• reflex vagal parasympathetic stimulation evoked by pain, gastric dilation, or fright
• ONLY 20% of patients with total high cord transection have neurogenic shock
21.
CLINICAL FINDINGS
Hypovolaemic shock
•COLD EXTREMITIES,
• REDUCED/ABSEBT PERIPHERAL
PULSES
• WEAK CENTRAL PULSES S/O LOW
CO
• DECREASE IN BP
• TACHYCARDIA
• TACHYOPNEA
• OLIGURIA
• CONFUSED ,LETHARGIC, ANXIOUS
• ALTERED SENSORIUM
CARDIOGENIC SHOCK
• OLIGURIA,
• ALTERED SENSORIUM, AND COLD
EXTREMITIES
• PERIPHERAL VASOCONSTRICTION
• TACHYCARDIA/DYSRHYTHMIAS
• DEPRESSED MENTAL STATUS
• SOME NEW ECG CHANGES LIKE
BUNDLE BLOCK AND T WAVE
ABNORMALITIES
• NECK VEINS DISTENSIONS
• SHOCK INDEX=HR/SBP >1.0 LV
DYSFUNCTION
INVESTIGATION
• Complete bloodcount
• Blood grouping and cross matching
• Serum electrolytes And lactates
• ABG Analysis
• Cardiac enzymes – rule out myocardial ischemia and diseases
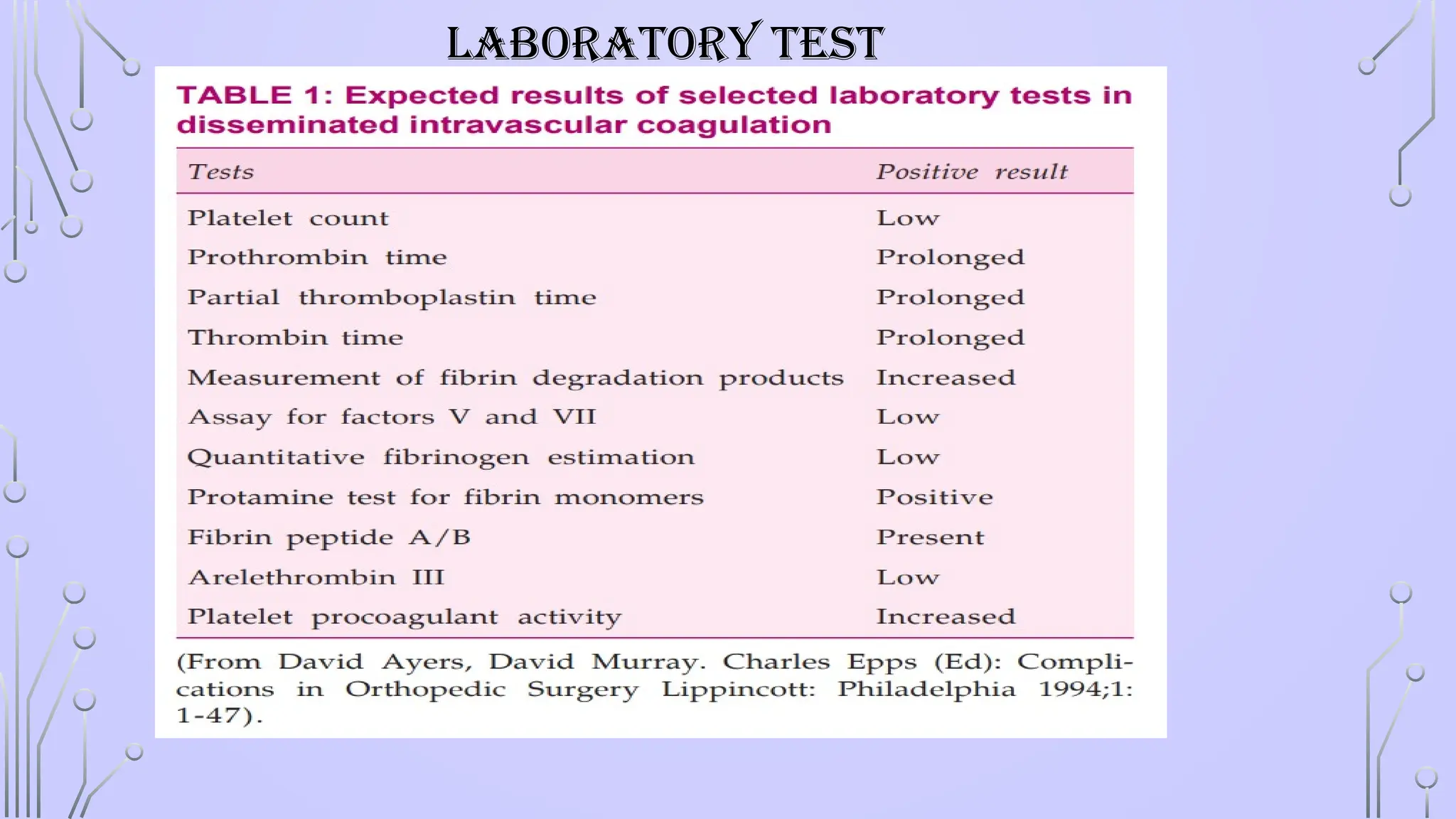
• Coagulation profile – PT,APTT,INR
• Chest xray, ECG , echocardiography
25.
TREATMENT
• Treatment dependsupon prompt assessment of the cause, type, severity and
duration of shock.
• Intial steps
• Assessments- bp, rr, pulses. Rbs etc.
• Secure airway,
• Oxygenation/ventilation
• Secure circulation -Two large bore iv cannulas,
• catheterisation – urine output rate less than 25ml/h suspected renal perfusion
• Position- Trendelenburg position / leg raised to 45 degree in supine position
• Control external haemorrhage – compression bandage
• Look for specific causes after stabilisation
26.
TREATMENT CONTD.
• Goalof treatment - restore safely intravascular volume and oxygen carrying
capacity
• Crystalloid first and then colloids / blood transfusion
• Fluid replacement- transfuse 1-2 ltsr of crystalloid
• Blood and blood products transfusion
• FRESH WHOLE BLOOD OR PACKED RBC IS IDEAL REPLACEMENT
• Ionotrophic agentsAND vasopressors – dopamine, dobutamine and non adrenaline
/ persistent hypotension
• Antibiotics – major role in septic shock after proper cultures report
• Antihistaminic and corticosteroids- in case of anaphylactic and septic shock
• Obstructive shock – tension pneumothorax-needle thoracotomy and chest tube
cardiac tamponade- pericardiocentesis, thoracotomy and chest tube
27.
END POINT OFRESUSTICATION ON SHOCK
When oxygen debt and acidosis are eliminated and aerobic metabolism restored,
treatment of the shock becomes successful.
. Normalization of blood pressure, HR and urine output
• Stabilized cardiac output: Cardiac index above 3L/min/m2 , mean arterial pressure
higher than or equal to 65 mm Hg, urine output more than 0.5 ml/kg/hr,
• Lactate and base deficit: Serum lactate less than 2 mM/L within 24 hours
• Restoration of aerobic metabolism: Systemic O2 consumption greater than 100
ml/min/m2 , mixed venous O2 saturation more than 70%
• Tissue pH (7.3 ± 0.1)
• Optimized oxygen delivery: Systemic O2 delivery more than 500 ml/min/m2 , SaO2
greater than 90%, Hb more than 7–9 gm/dL.
• Restored hemostasis: INR < 1.5, aPTT < 1.5 × control, platelet count > 50 × 109/L
28.
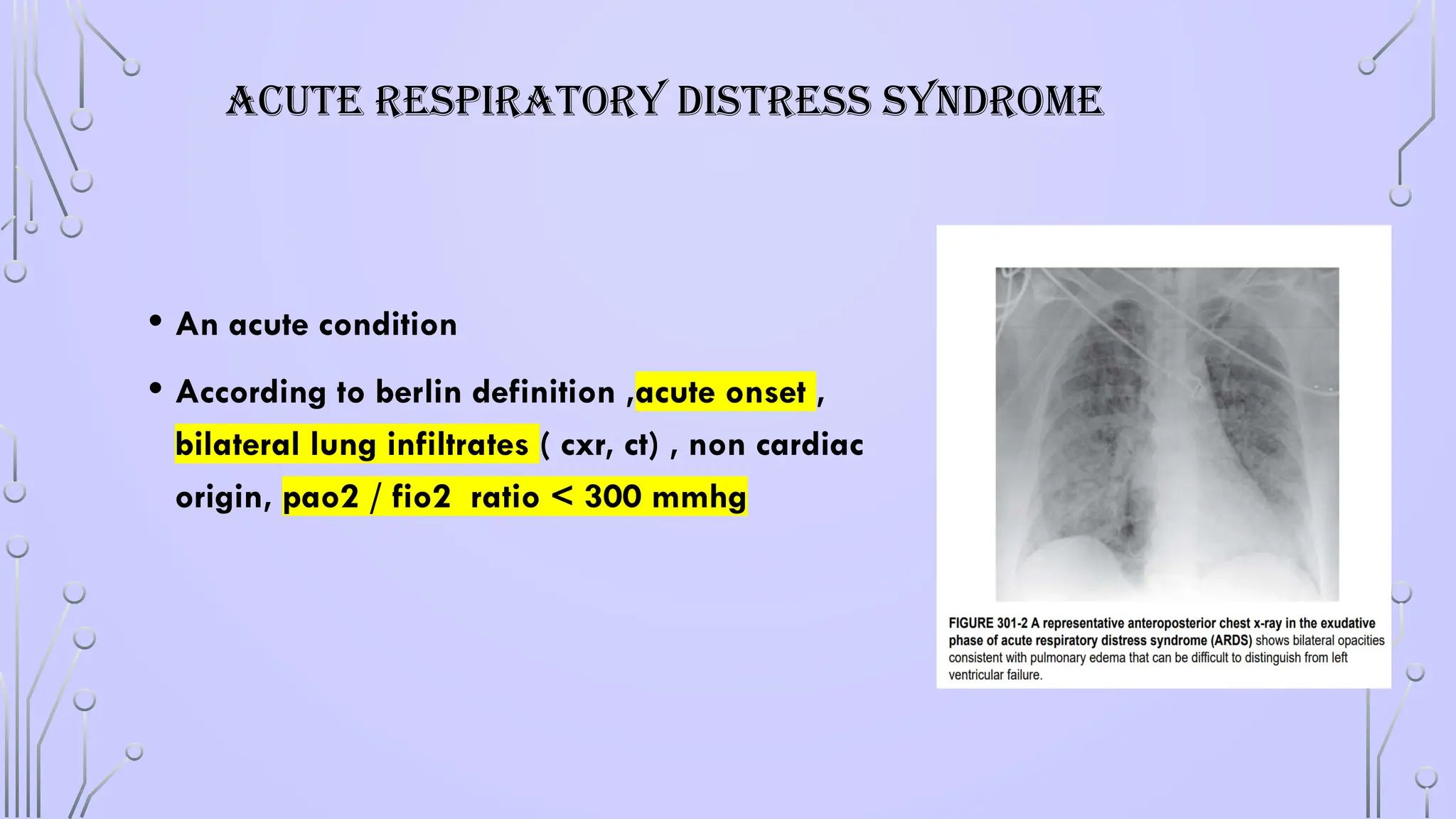
ACUTE RESPIRATORY DISTRESSSYNDROME
• An acute condition
• According to berlin definition ,acute onset ,
bilateral lung infiltrates ( cxr, ct) , non cardiac
origin, pao2 / fio2 ratio < 300 mmhg
29.
EPIDEMIOLOGY OF ARDS
•Incidence - 64.2 to 78.9 cases per 1lakh person
• 10 to 15 % of ICU patients and up to 23 % of mechanically
ventilated person meet the criteria of ARDS
30.
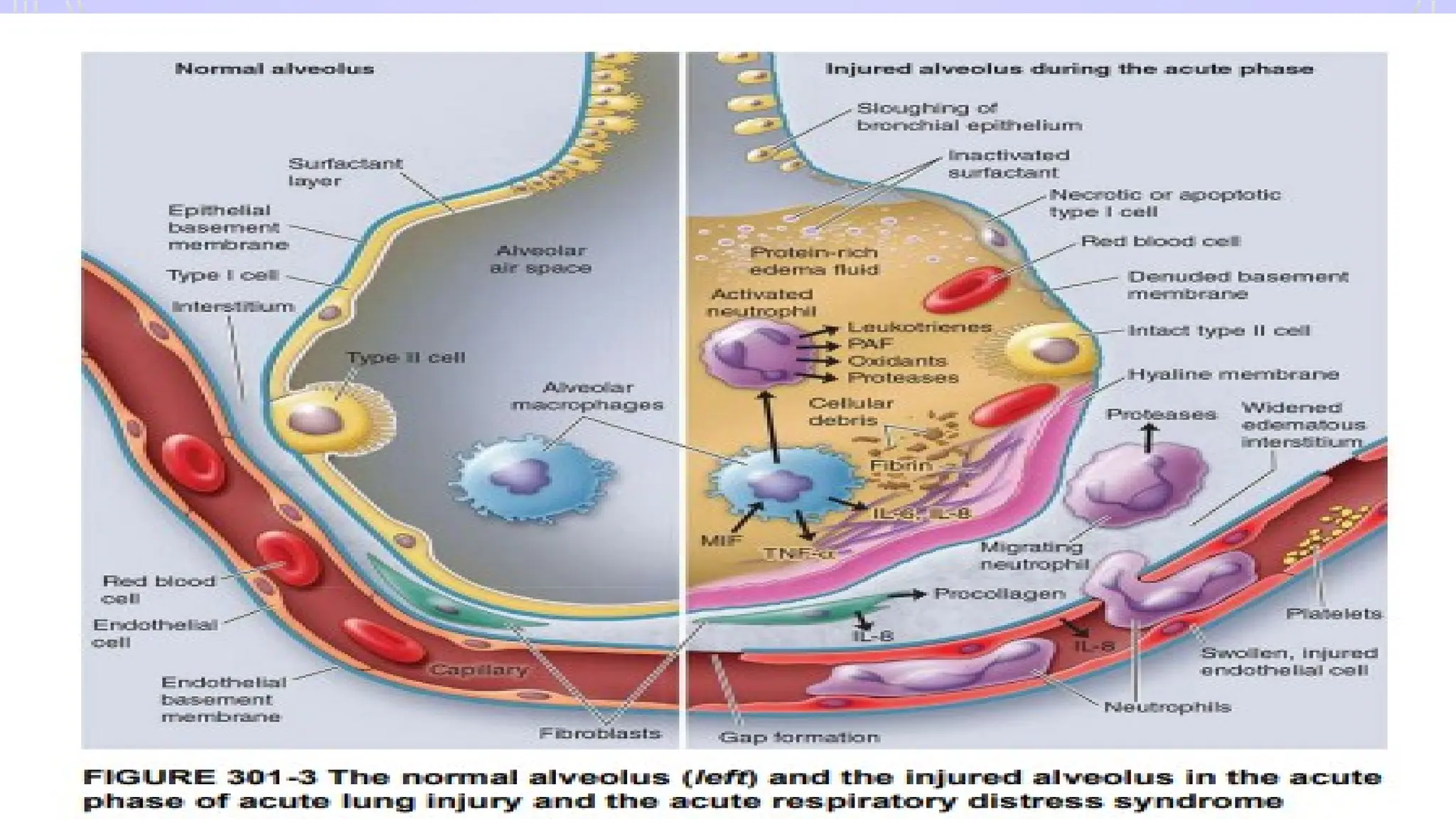
PATHOPHYSIOLOGY- ARDS
• Diffusealveolar damage and lung capillary endothelial injury
• 2 phase
• Early- exudative
• inc. permeability of alveoli capillary barrier, influx of fluid into
alveoli
• Late- fibroproliferative in character, IL-1 key mediator
• pulmonary fibrosis– neovascularisation and accumulation in
alveolar spaces
32.
ETIOLOGY- ARDS
• Sepsis
•Trauma, with or without pulmonary contusion
• Massive transfusion
• Bacteraemia
• Fractures, particularly multiple fractures and long bone fr
• Pneumonia
• Drug overdose
• Post-perfusion injury after cardiopulmonary bypass
• Fat embolism
• Aspiration
33.
CLINICAL PRESENTATION ANDINVESTIGATION- ARDS
• Acute onset of dyspnea
• rapid shallow breathing and hypoxemia
• Feeling of not able to get enough air to breathe
• Tachypnoea and tachycardia
• Arterial blood gas – arterial hypoxemia. Severity
• Mild: 200 mm hg < pao2 /fio2 < 300 mm hg
• Moderate: 100 mm hg < pao2 /fio2 < 200 mm hg
• Severe: pao2 /fio2 < 100 mm hg
• CHEST XRAY- BILATERAL ALVEOLAR and interstitial infiltrates
• Absence of left atrial hypertension- pcwp< 18mmhg
DIFFERENTIATE ARDS FROM CARDIOGENIC PULMONARY OEDEMA
Diagnostic
criteria
34.
TREATMENT- PRINCIPLE INARDS
• SUPPORTIVE CARE
• FOCUS ON REDUCING SHUNT FRACTION
• INCREASING O2 DELIVERY
• REDUCING O2 CONSUMPTION
35.
TREATMENT
• Identification andtreatment of cause
• Mechanical ventilation- –
• LUNG PROTRECTIVE VENTILATOR STRATEGY
• high peep
• Oxygenation
• Fluid management
• Neuromuscular blockade-synchrony to mechanical ventilation
• glucocorticoids
36.
CRUSH SYNDROME /TRAUMATICRHABDOMYOLYSIS
• Due to prolonged continuous pressure on muscle tissue.
• Seen victims who are rescued from beneath rubble after several
hours or days of entrapment.
• Patients of drug addiction who have compressed their own
extremity
37.
PATHOPHYSIOLOGY – CRUSHSYNDROME
• impairment of sarcolemmic sodium-potassium-adenosine triphosphate activity.
• pressure is released, the metabolics accumulated in the ischemic area are
released into circulation.
• Large amount of intracellular potassium, phosphorus, lactic acid and myoglobin
are released into the circulation.
• Fluid shifts can produce shock
• . Renal failure results in acidosis.
• Hyperkalemia, hyperphosphatemia, hypocalcemia, myoglobinuria, and
metabolic acidosis may begin within hours of rescue in the extricated and
untreated patient.
38.
TREATMENT- CRUSH INJURY
•Emergency treatment should be started with saline infusion
• When a urine flow has been established, a forced mannitol-alkaline diuresis of
up to 8 l/d should be maintained (urine pH greater than 6.5).
• Alkalinization increases the urine solubility of acid hematin and aids in its
excretion.
• continued until myoglobin no longer is detectable in the urine.
• Mannitol also removes oxygen-free radicals.
• Allopurinol - limiting the reperfusion injury by inhibiting xanthine oxidase
activity protection.
• Renal failure generally can be averted with the aggressive treatment.
39.
DIC-DISSEMINATED INTRAVASCULAR
COAGULATION
• Disseminatedintravascular coagulation is characterized by consumption
of coagulation factors and increased fibrinolytic activity that leads to
excessive bleeding.
• DIC develops in those situations in which coagulation system is
stimulated.
• causes of DIC-the most common causes are FAT EMBOLISM, SEPSIS,
TRAUMA, etc.
40.
TYPES OF DIC
Acuteor decompensated DIC:
• rapid and extensive activation of coagulation leading to significant bleeding
• consumption of coagulation factors and widespread microvascular thrombosis with
• consequent end-organ damage.
• Examples are sepsis and trauma.
Chronic or compensated DIC:
• slow activation of coagulation with slow consumption of coagulation factors.
• coagulation factor levels are normal or increased as they are replenished.
• clinical features are minimal or absent and laboratory abnormalities are the only
evidence of DIC.
• Examples :intrauterine retention of dead foetus, liver disease, giant haemangioma,
eclampsia, and malignancy.
41.
PATHOPHYSIOLOGY OF DIC
•Triggering event- activation of monocytes and endothelial cells-
• generate tissue factor in cell surface- activation of coagulation cascade
• Abundant intravascular thrombin , increases fibrinogen to fibrin
conversion
• Widespread fibrin and platelet deposition in capillaries and arterioles
42.
PATHOPHYSIOLOGY CONTD.
• Thrombosis, multi organ failure
• Depressed clotting inhibitory mechanism ( dec . antithrombin iii and
protein c)
• Excessive clotting activates fibrinolytic system
• Breaking of clots, increases FDP inhibits normal blood clotting
• Blood loses ability to clot , haemorrhage
43.
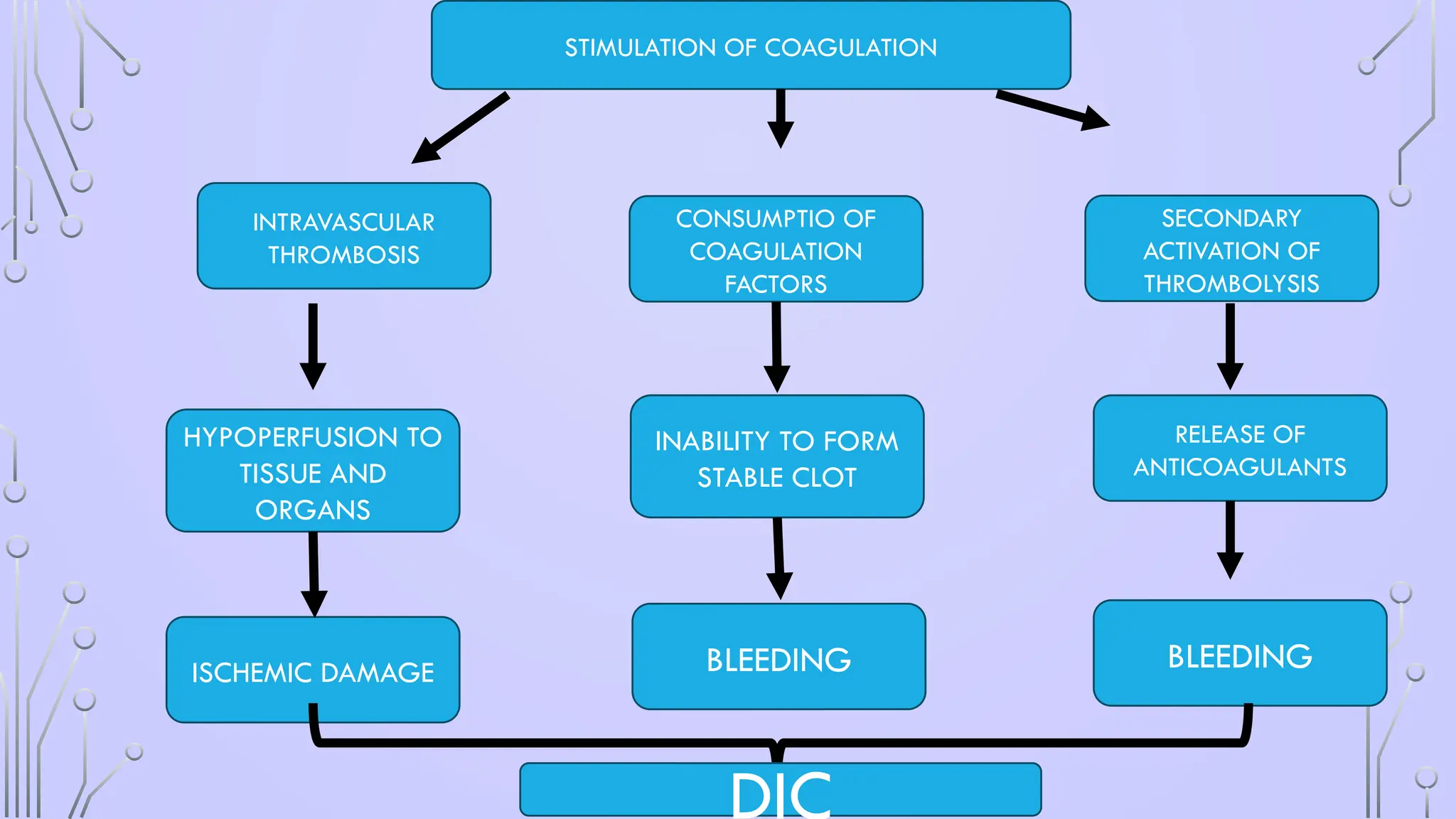
BLEEDING
INABILITY TO FORM
STABLECLOT
CONSUMPTIO OF
COAGULATION
FACTORS
BLEEDING
RELEASE OF
ANTICOAGULANTS
SECONDARY
ACTIVATION OF
THROMBOLYSIS
HYPOPERFUSION TO
TISSUE AND
ORGANS
ISCHEMIC DAMAGE
INTRAVASCULAR
THROMBOSIS
STIMULATION OF COAGULATION
DIC
44.
CLINICAL PRESENTATIONS
• BLEEDING– Any site, petechiae, bruises , haematoma,
Ecchymoses, Mostly in acute DIC
• THROMBOSIS- Digital ischaemia and gangrene,
Mostly in chronic DIC
• HYPOTENSION OR SHOCK
• ORGAN DYSFUNCTION- Cerebral involvement is characterized by
convulsions, coma and mental changes
ANTICOAGULANT THERAPY
• HEPARIN:Heparin therapy should be started to prevent microthrombi.
• frequently monitored with fibrinogen estimation, platelet count and clinical assessment
• . Usually, an infusion of heparin 8 to 15 units/kg/hour is often successful.
• ANTI THROMBIN III
• TRANEXAMIC ACID
• HIRUDIN
• EPSILON AMINO CPROIC ACID
#3 Trauma is a major cause of death and disability worldwide that mainly affects young adults.
50% of fatally injured casualties died from non-survivable injuries immediately, or within minutes after the accident; 30% survived the initial trauma, but died within 1–3 hours; the remaining 20% died from complications at a late stage during the 6 weeks after injury (
#4 initial mortality peak is usually due to nonsurvivable, central nervous system injury or cardiovascular disruption
e second peak of deaths during the first few hours after injury is most often due to hypoxia and hypovolaemic shock. A significant proportion of these deaths can be avoided with an effective emergency medical service (EMS), as has been demonstrated in the UK since 2012; hence, this period has been called ‘the golden hour’.
e third peak in the cumulative mortality rate within the 6 weeks following injury was largely due to multisystem failure and sepsis
#5 physiologic response to injury 3 phases:
(a) a hypodynamic ebb phase (shock) ---- body initially attempts to limit the blood loss to maintain perfusion to the vital organs;
(b) a hyperdynamic flow phase lasting for up to 2 weeks, increased blood flow, in order to remove waste products and to allow nutrients to reach the site of injury for repair; and
(c) a recuperation phase, lasting for months, to allow the human body to attempt to return to its pre-injury level.
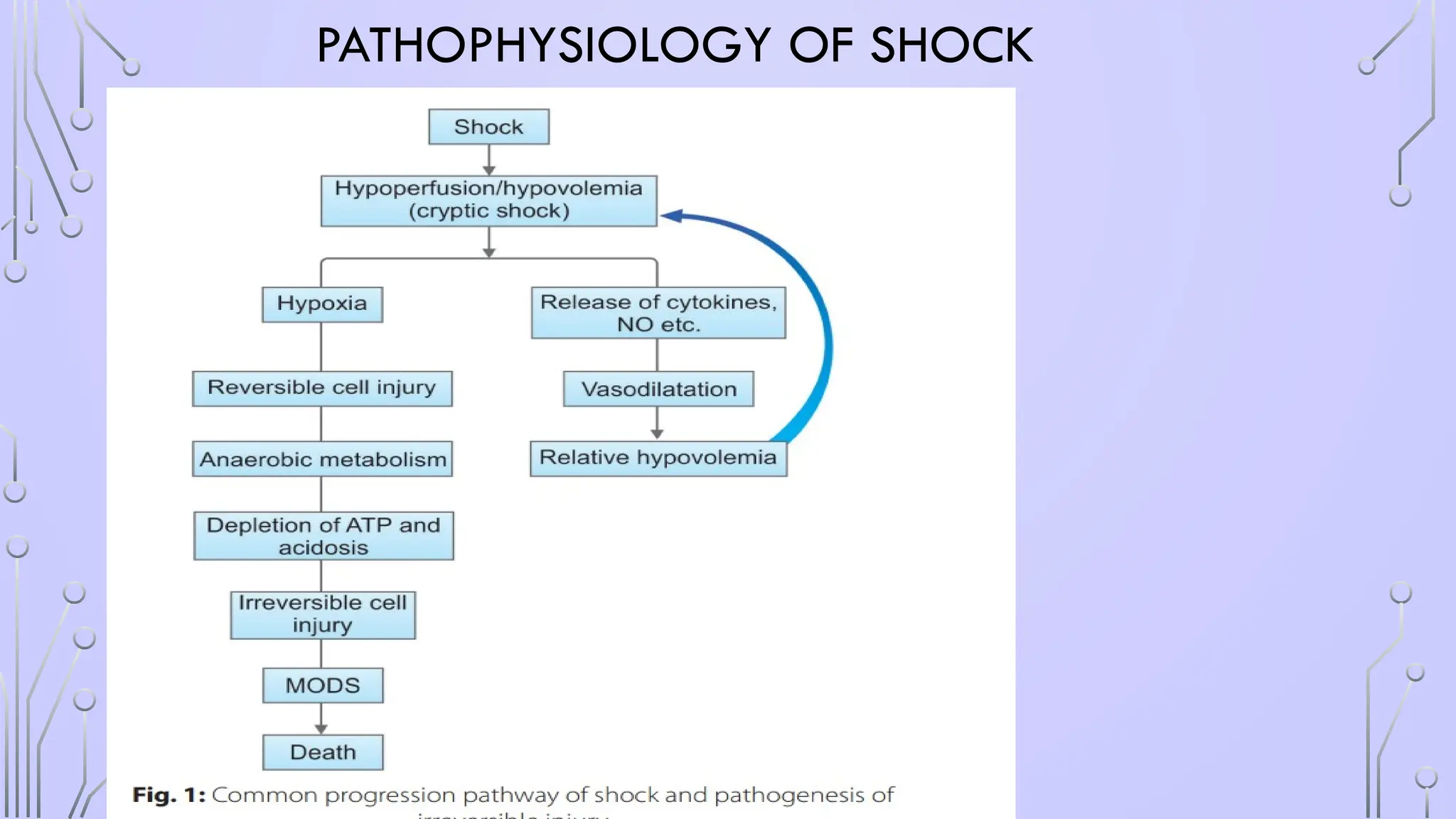
#6 A fracture is associated with damage to bone, periosteum, and adjacent soft tissues such as muscle and connective tissue. Adjacent blood vessels bleed into the affected area and cause a hematoma.
Blood loss and tissue damage caused by fractures and soft tissue crush injuries induce generalized hypoxemia in the entire vascular bed of the body. Hypoxemia is the leading cause of damage as it causes all endothelial membranes to alter their shape. Subsequently, the circulating immune system, namely the neutrophil and macrophage defense systems, identify these altered membranes.
sequestration and the activation mainly of the polymorphonuclear granulocytes (PMN), the monocytes, and the lymphocytes trigger a multifocal molecular and pathophysiologic process
The cells interact and adhere to the endothelium via adhesion molecules like L-selectin, ICAM-1, and integrin β2 (representatives of the selectin, immunoglobulin, and integrin superfamilies, respectively). After firm adhesion, PMN leukocytes can extravasate and by losing their autoregulatory mechanisms can release toxic enzymes causing remote organ injury in the form of ARDS, MODS.127,15
The sequestration and the activation mainly of the polymorphonuclear granulocytes (PMN), the monocytes, and the lymphocytes trigger a multifocal molecular and pathophysiologic process
#9 defining feature of shock is tissue hypoperfusion, and not a predetermined level of systemic arterial blood pressure
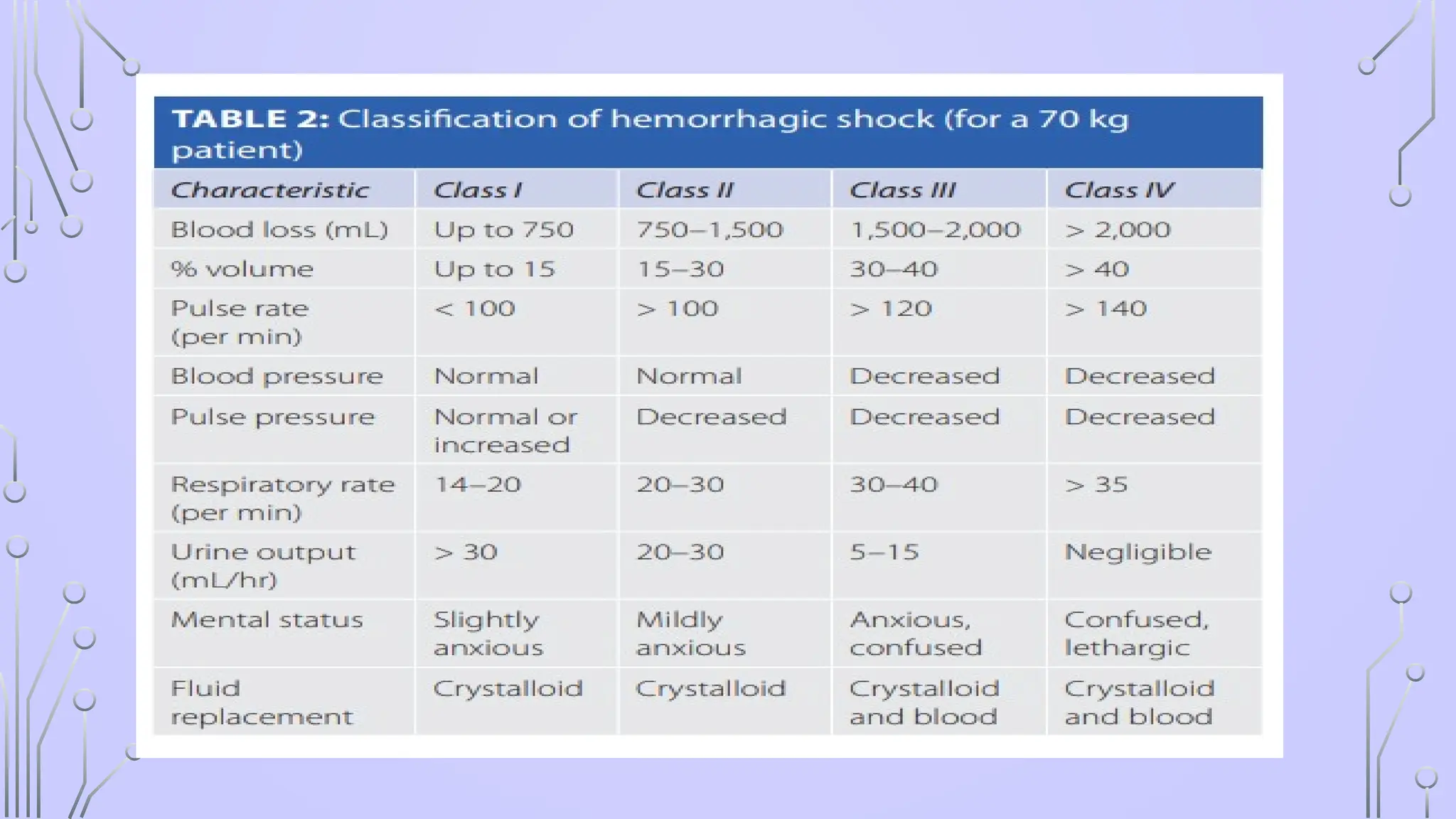
#12 HEMORRHAGIC HYPOVOLEMIA : m/c cause of shock in trauma ,
decreased cardiac output leading to hypoperfusion of the tissues and cell death.
NONHEMORRHAGIC” HYPOVOLEMIC SHOCK- massive gastrointestinal (GI) fluid or urinary losses leading to severe dehydration (as in diabetic ketoacidosis or diarrhea from cholera).
#18 Sepsis” is defined by presence of at least two of the four signs of the systemic inflammatory response syndrome (SIRS, see Chapter 22) along with documented signs of infection: 1. Temperature higher than 38°C and less than 36°C 2. Tachycardia (> 90 beats/minute) 3. Tachypnea (> 20 breaths/minute), hypocapnia (partial pressure of carbon dioxide < 32 mm Hg), or the need for mechanical ventilatory assistance and 4. Leukocytosis (> 12,000 cells/mm3 ), leukopenia (< 4,000 cells/mm3 ), or a left shift (> 0% immature band cells) in the circulating white cell differential. “Bacteremia” is defined as the growth of bacteria from blood cultures. – “Severe sepsis” is sepsis associated with dysfunction of one or more organ systems (e.g. altered sensorium, hypoxia, decreased urine output). Add this to hypotension and it gives “septic shock”
#20 incomplete motor or sensory deficits (or both) rarely have hypotension
suspected in all hypotensive patients of trauma without any evidence of active hemorrhage.
Psychogenic or neurogenic factors such as spinal cord injury, trauma pain, gastric dilation may produce reflex vegal stimulation with decreased cardiac output, hypotension and decreased cerebral blood flow
#22 TO MAKE THE DIAGNOSIS, IT IS IMPORTANT TO DOCUMENT MYOCARDIAL DYSFUNCTION
History of pre-existing cardiac disease or chest trauma is present
#26 P. For patients with all types of shock, there can be development of ARDS and subsequent V̇ /Q̇ mismatch and shunt. Supplemental oxygen should be initiated and titrated to maintain SpO2 of 92–95%. This may require intubation and initiation of mechanical ventilation.
#27 Norepinephrine is the first-choice vasopressor, with potent α1 and β1 adrenergic effects. The α1 causes vasoconstriction while β1 has positive inotropic and chronotropic effects.
#28 higher serum lactate and higher base deficits predicts MODS
#29 bilateral pulmonary infiltrates and severe hypoxemia in the absence of evidence for cardiogenic pulmonary edema.”
ARDS can be simply defined as non-cardiogenic pulmonary edema.











































![APPROACH TO SHOCK [Auto-saved].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/approachtoshockauto-saved-230707035943-4dc369a3-thumbnail.jpg?width=640&height=640&fit=bounds)

























































