Downloaded 20 times











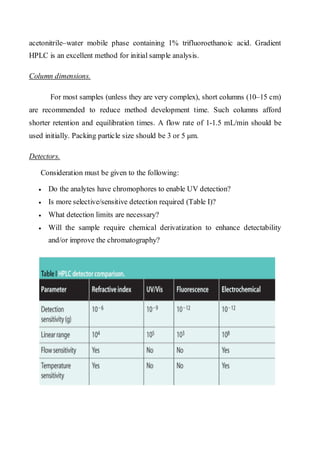


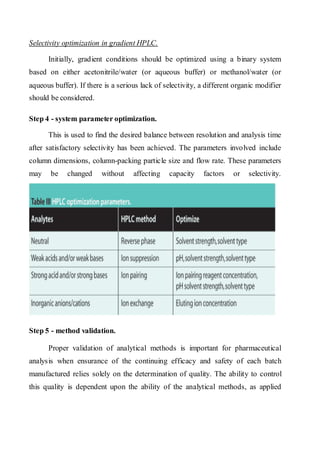











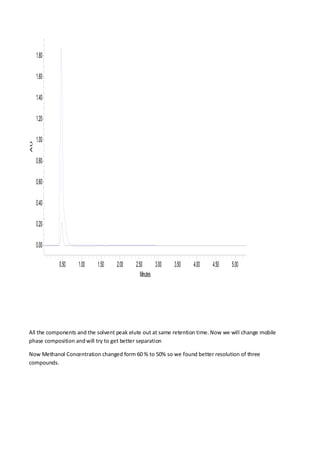
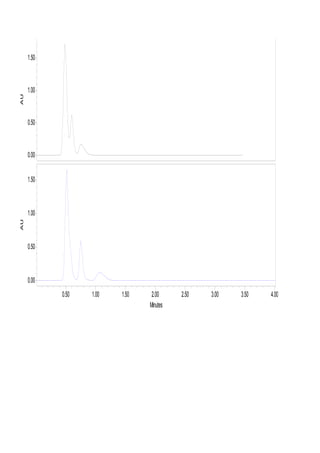
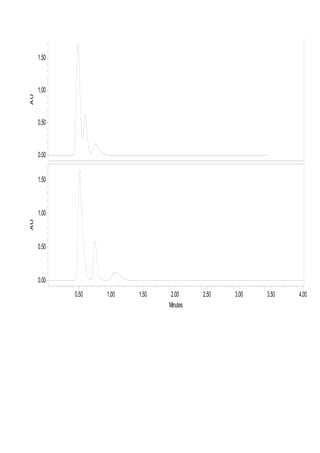
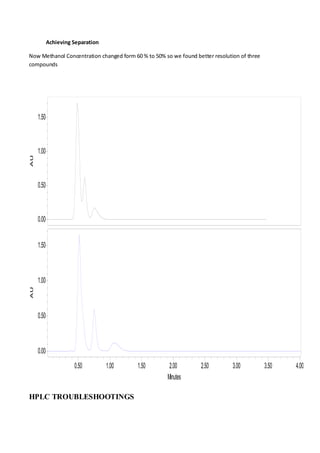

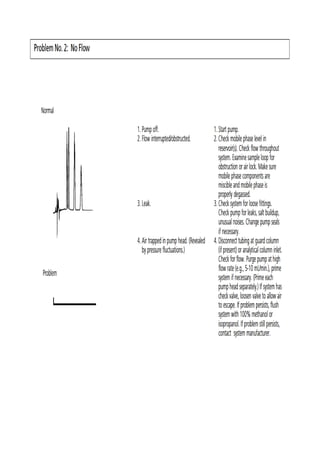
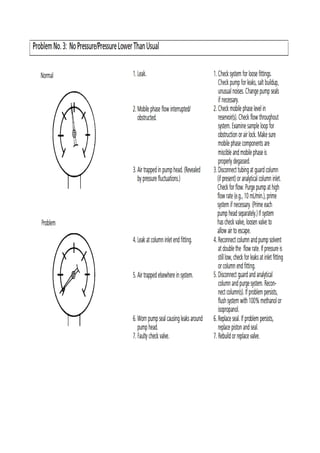
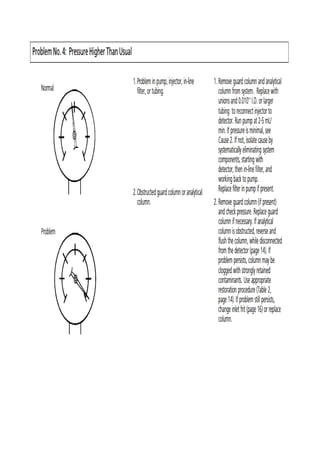
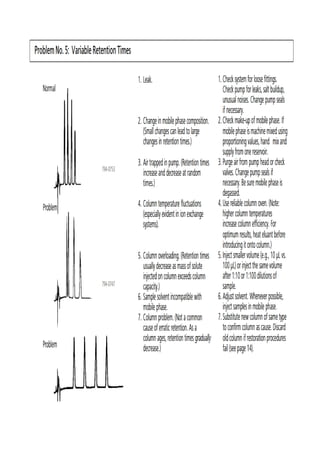
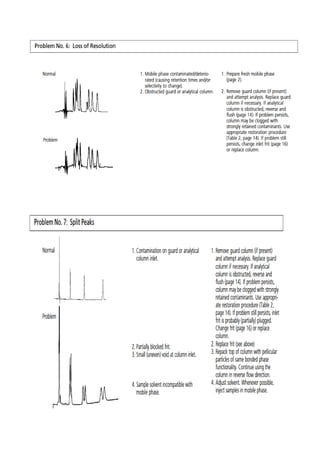

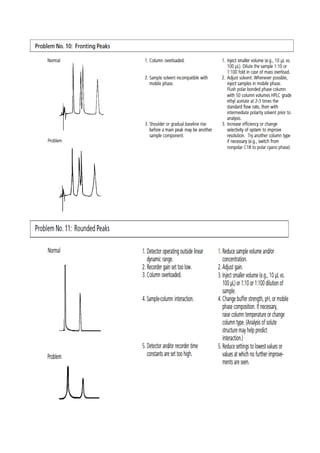


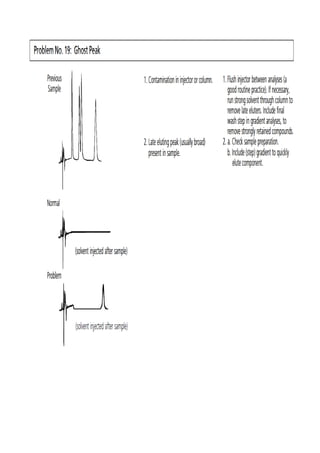







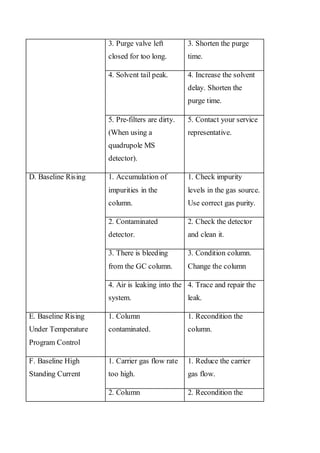
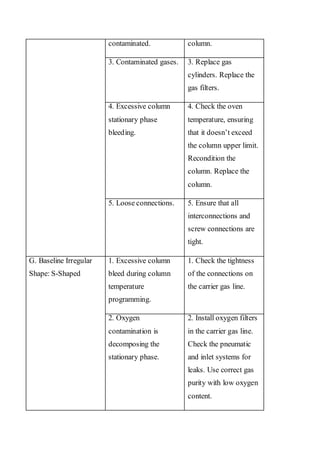


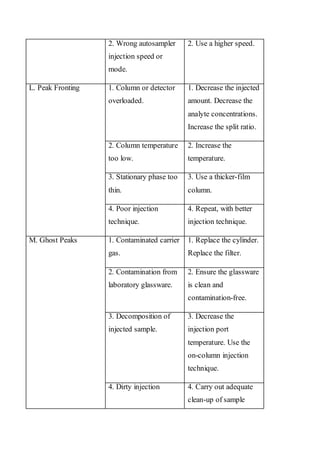






Liquid chromatography uses two immiscible liquid phases to separate components of a mixture. The stationary phase is adsorbed to a support and the mobile phase flows through, carrying solutes between the phases. Separation occurs as solutes distribute differently between the phases based on their properties. Liquid-solid chromatography uses a solid stationary phase, like silica or alumina, which can selectively retain solutes based on interactions between functional groups. Quantitative analysis with chromatography requires optimizing the separation, identifying peaks, establishing a calibration curve to determine detection limits and linearity, and validating the method to ensure consistent, accurate results.











































































































