Downloaded 816 times








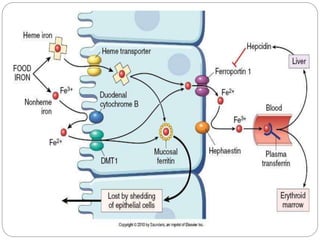

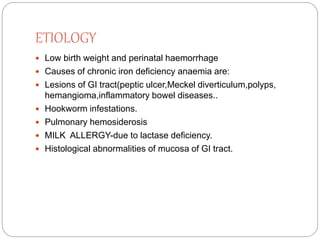


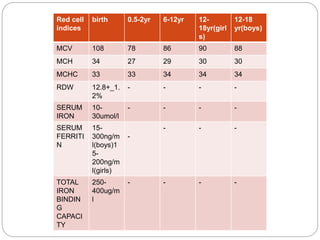

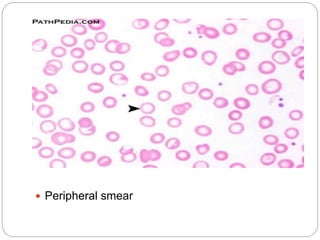






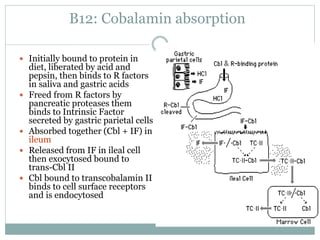
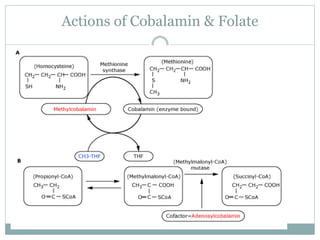



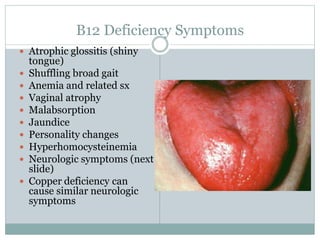

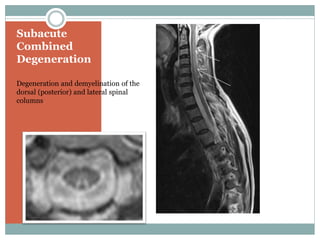
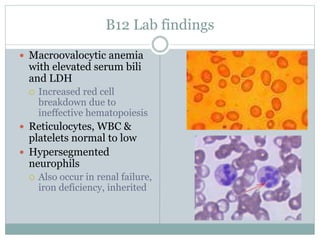
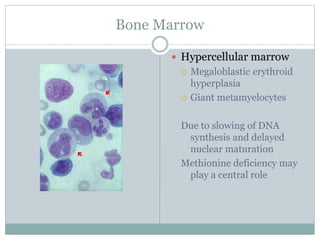

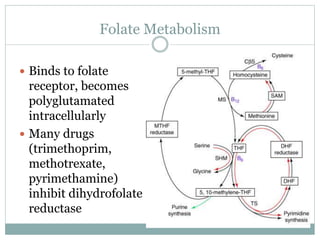



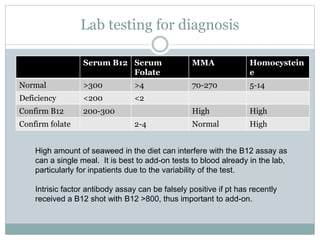
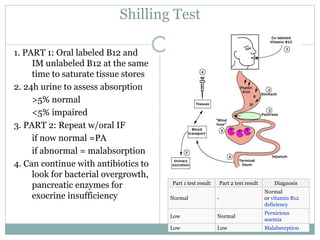





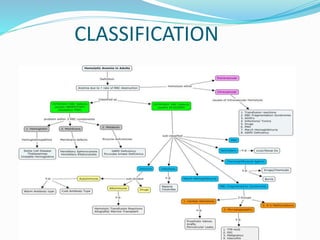

































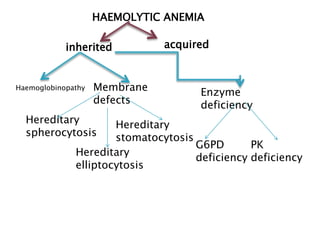

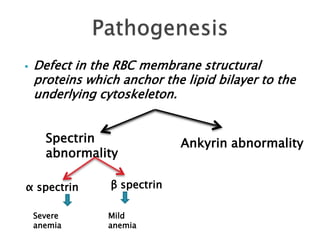
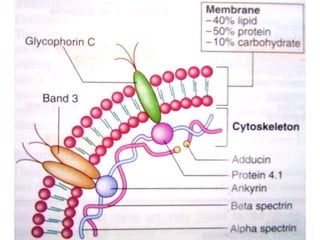


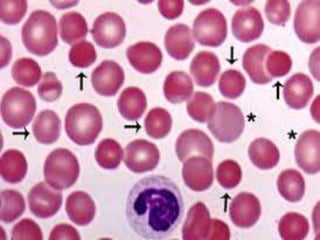
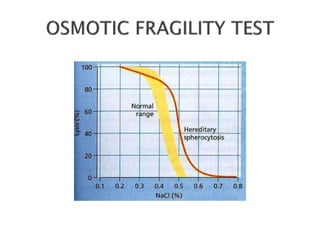


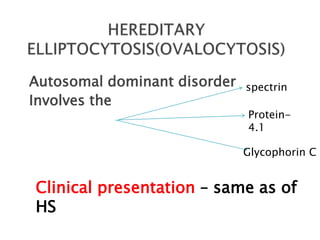
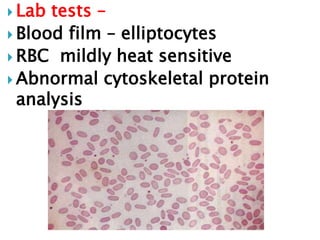


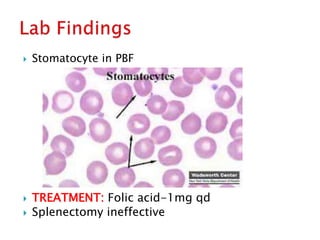

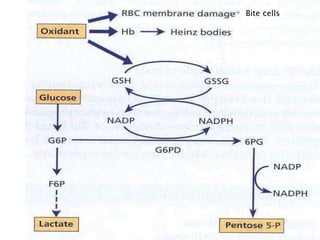


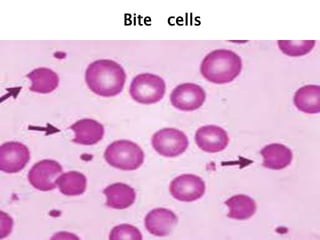






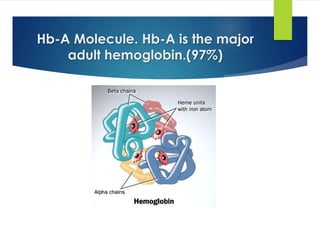


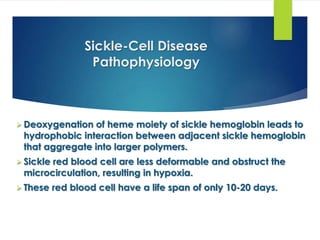




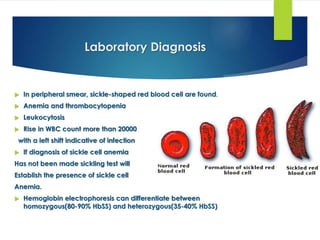














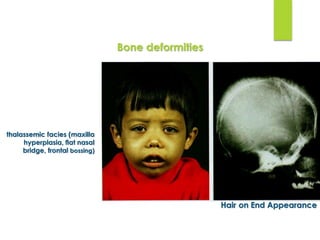





This document provides definitions and information about anemia and iron deficiency anemia. It begins by defining anemia based on hemoglobin and hematocrit levels below certain thresholds. It then classifies anemias based on pathophysiology and morphology. Iron deficiency anemia is discussed in depth, including iron metabolism, sources of iron, clinical manifestations, investigations, and management with oral or parenteral iron supplementation or blood transfusions. Megaloblastic anemia is then introduced, focusing on vitamin B12 and folate, causes of B12 deficiency including pernicious anemia and effects of aging, and symptoms of B12 deficiency including neurological effects.























































































