Download as PDF, PPTX


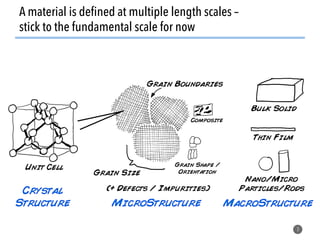
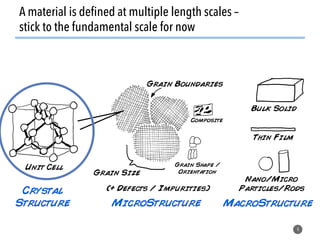


![What constrains traditional experimentation?
7
“[The Chevrel] discovery resulted from a lot of
unsuccessful experiments of Mg ions insertion
into well-known hosts for Li+ ions insertion, as
well as from the thorough literature analysis
concerning the possibility of divalent ions
intercalation into inorganic materials.”
-Aurbach group, on discovery of Chevrel cathode
for multivalent (e.g., Mg2+) batteries
Levi, Levi, Chasid, Aurbach
J. Electroceramics (2009)](https://image.slidesharecdn.com/citrine-180703163402/85/High-throughput-computation-and-machine-learning-methods-applied-to-materials-design-7-320.jpg)









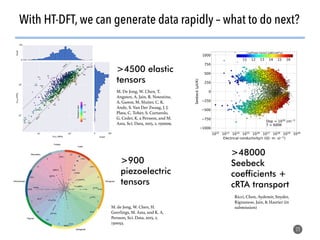

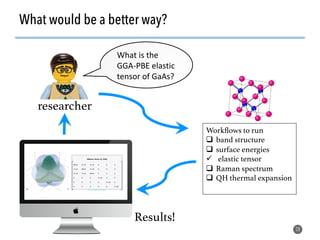

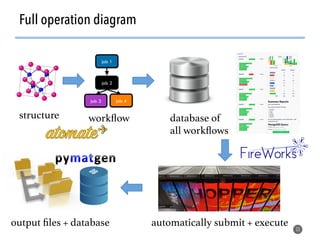





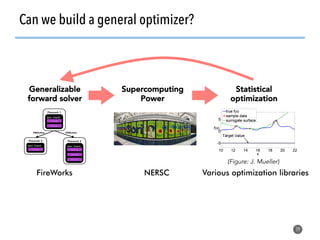
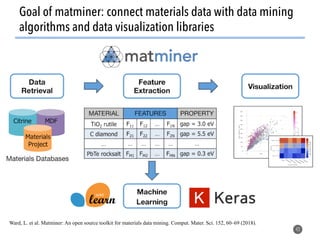
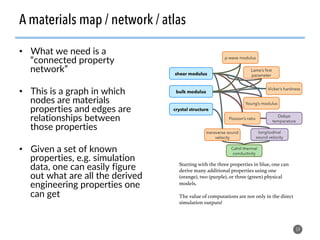
![>40 featurizer classes can
generate thousands of
potential descriptors
43
Matminer contains a library of descriptors for various
materials science entities
feat = EwaldEnergy([options])
y = feat.featurize([input_data])
• compatible with
scikit-learn
pipelining
• automatically deploy
multiprocessing to
parallelize over data
• include citations to
methodology papers](https://image.slidesharecdn.com/citrine-180703163402/85/High-throughput-computation-and-machine-learning-methods-applied-to-materials-design-43-320.jpg)

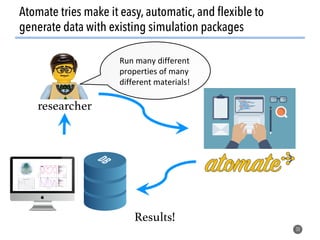
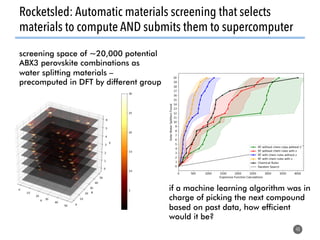
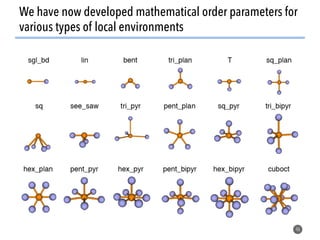
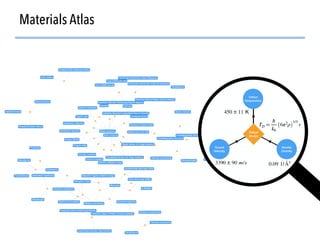
![Defining local order parameters for various environments
45
Use a given local order parameter
with a threshold
for motif recognition:
If qtet > qthresh,
then motif is tetrahedron.
Else
not (too much) a tetrahedron.
Tetrahedral order parameter, qtet, [1]:
[1] Zimmermann et al., J. Am. Chem. Soc., 2017, 10.1021/jacs.5b08098](https://image.slidesharecdn.com/citrine-180703163402/85/High-throughput-computation-and-machine-learning-methods-applied-to-materials-design-45-320.jpg)
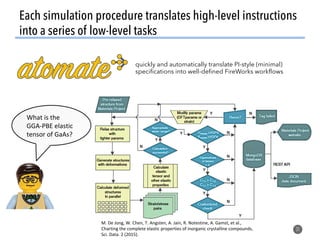
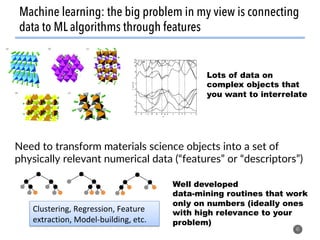
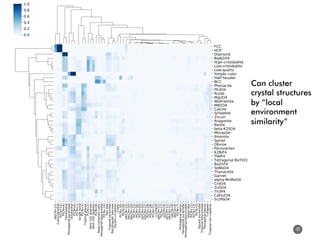
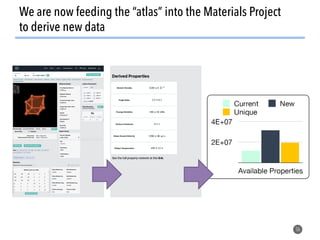
![How well do these work?
47
1. Order parameters clearly
distinguish different environments
even after thermal distortion
2. Work well in applications (defect site
finding, diffusion characterization)
[1] Zimmermann et al., Frontiers of Materials, 2017, doi: 10.3389/fmats.2017.00034](https://image.slidesharecdn.com/citrine-180703163402/85/High-throughput-computation-and-machine-learning-methods-applied-to-materials-design-47-320.jpg)

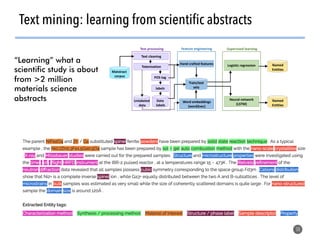




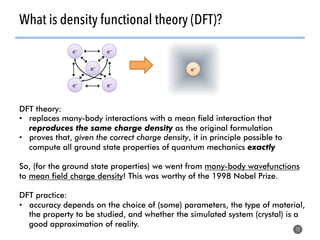
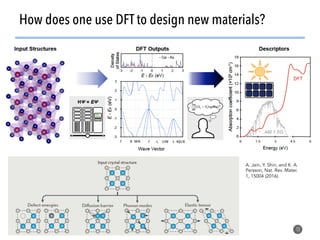
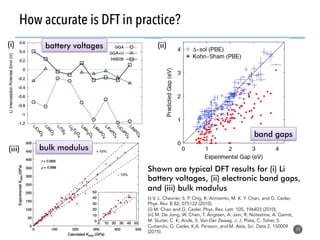
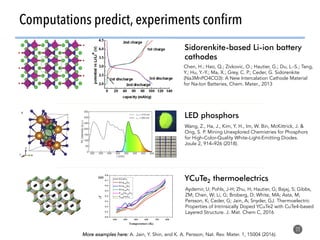
High-throughput computation and machine learning methods can be applied to materials design problems at scale. Density functional theory (DFT) allows modeling of materials at the quantum mechanical level but large computational resources are required. "High-throughput DFT" uses automation, parallelization across supercomputers, and data mining approaches to rapidly screen millions of potential new materials in silico before experimental validation. This helps address the challenge of discovering new materials for applications like energy technologies by searching the vast space of possible compositions and structures more efficiently than traditional experimentation alone.























































































