
Downloaded 68 times
![SEMINAR ON
IPQC, RELEASE OF FINISHED PRODUCTS
PRESENTED BY
HUZAIFA NAAZ
H.T.NO.636217885002
M.Pharmacy I year
UNDER GUIDANCE OF
DR. SANDIP SEN
DEPARTMENT OF PHARMACEUTICAL ANALYSIS
SRIKRUPA INSTITUTE OF PHARMACEUTICAL SCIENCES
[Affiliated to Osmania university]
[Approved by PCI; AICTE]](https://image.slidesharecdn.com/qaandqcseminar-180705183724/85/Qa-and-qc-seminar-1-320.jpg)
![SEMINAR ON
IPQC, RELEASE OF FINISHED PRODUCTS
PRESENTED BY
HUZAIFA NAAZ
H.T.NO.636217885002
M.Pharmacy I year
UNDER GUIDANCE OF
DR. SANDIP SEN
DEPARTMENT OF PHARMACEUTICAL ANALYSIS
SRIKRUPA INSTITUTE OF PHARMACEUTICAL SCIENCES
[Affiliated to Osmania university]
[Approved by PCI; AICTE]](https://image.slidesharecdn.com/qaandqcseminar-180705183724/75/Qa-and-qc-seminar-1-2048.jpg)









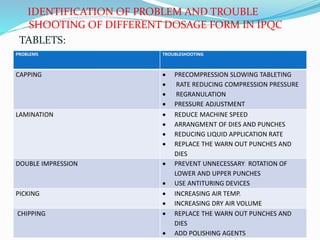
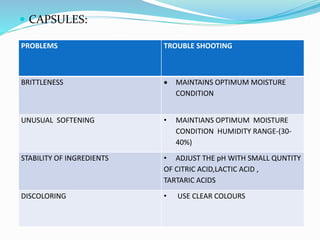


















The document summarizes the key aspects of in-process quality control (IPQC), release of finished products, and related quality control procedures. It defines IPQC and discusses the various tests involved at different stages of production. These include physical, chemical, biological, and microbiological testing. It also outlines the documentation required for batch release, including quality review, auditing, and maintaining distribution records. The presentation provides an overview of quality control measures from raw material receipt through finished product release.


















































































