Downloaded 136 times








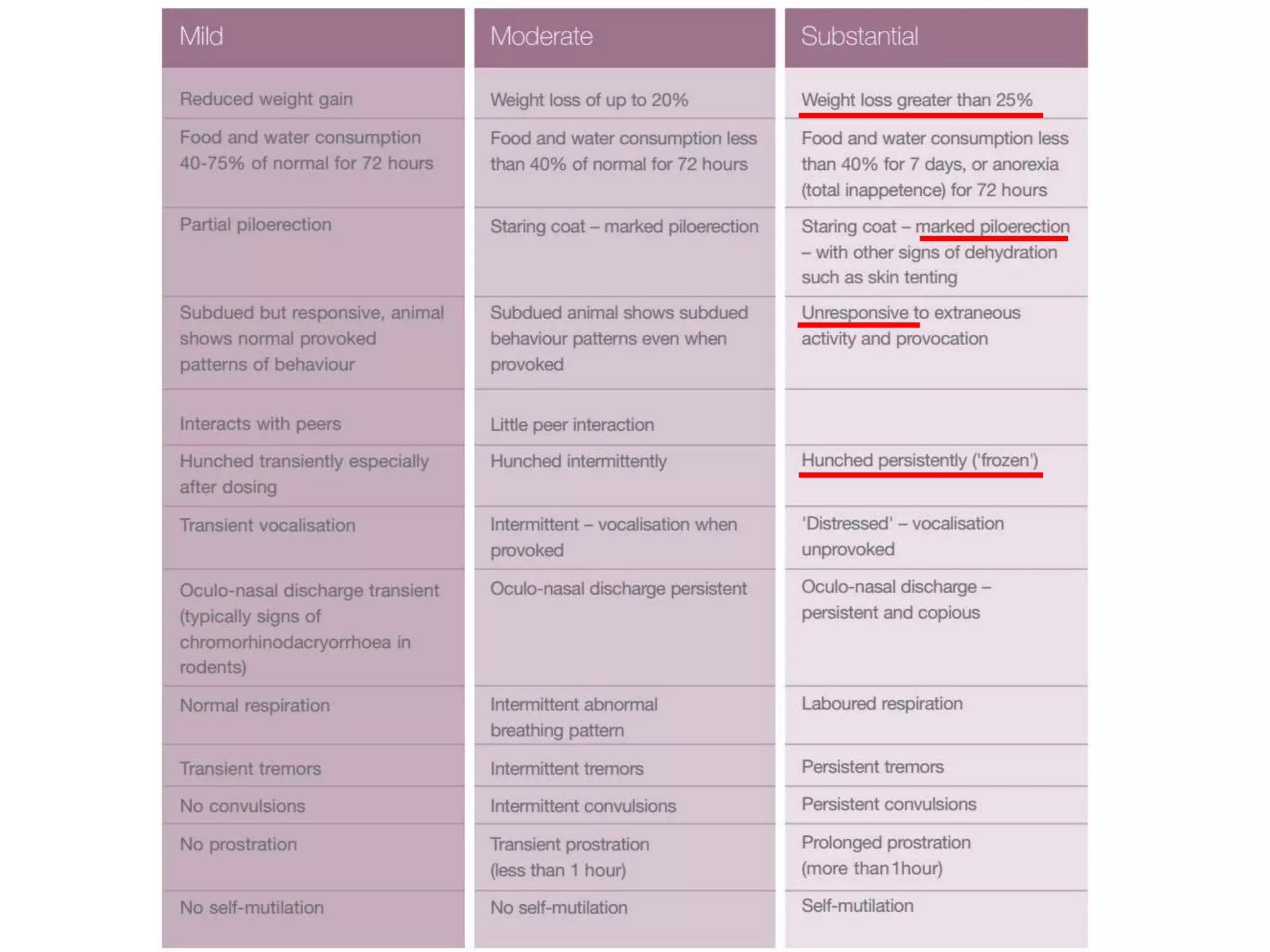








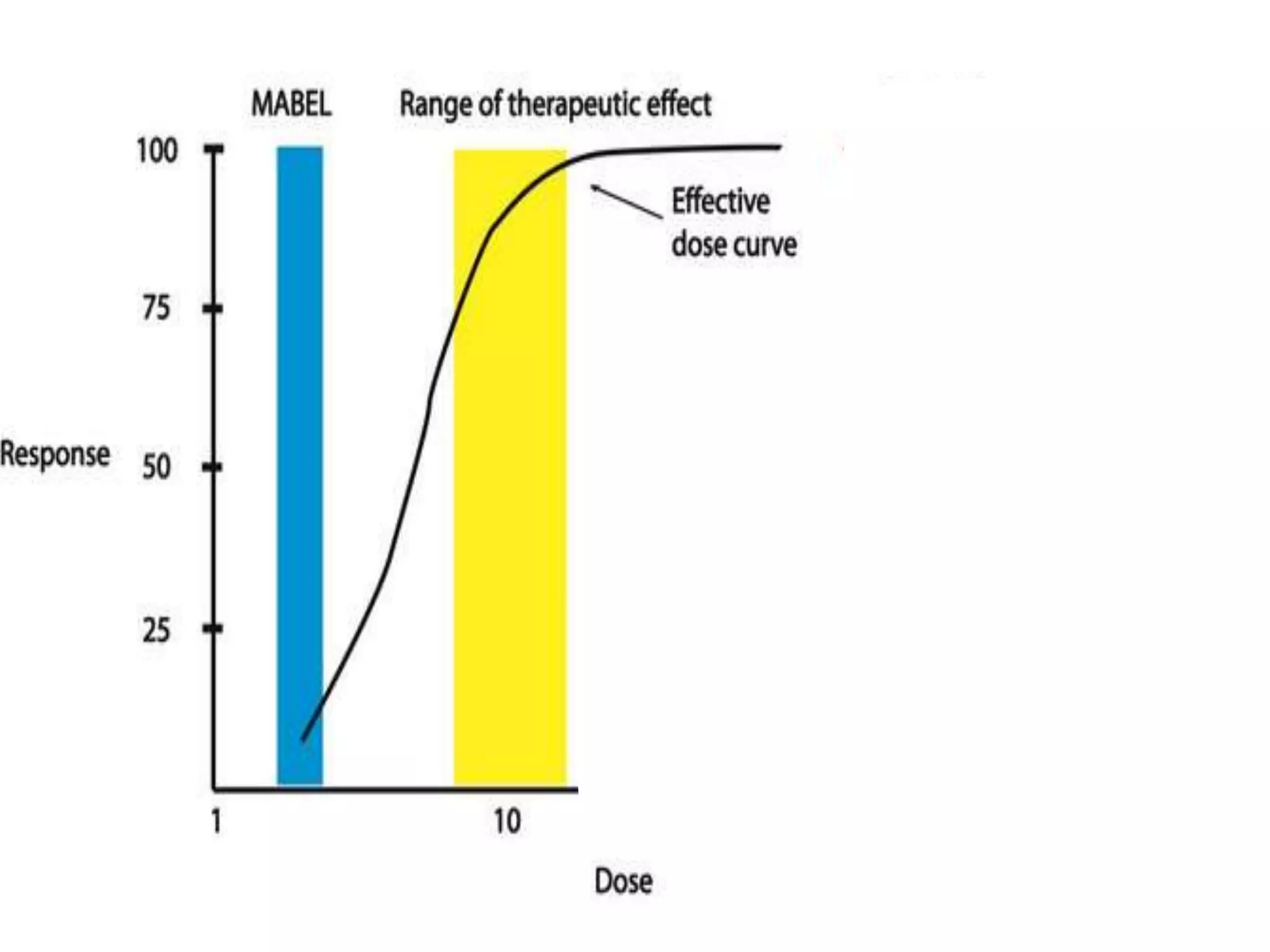
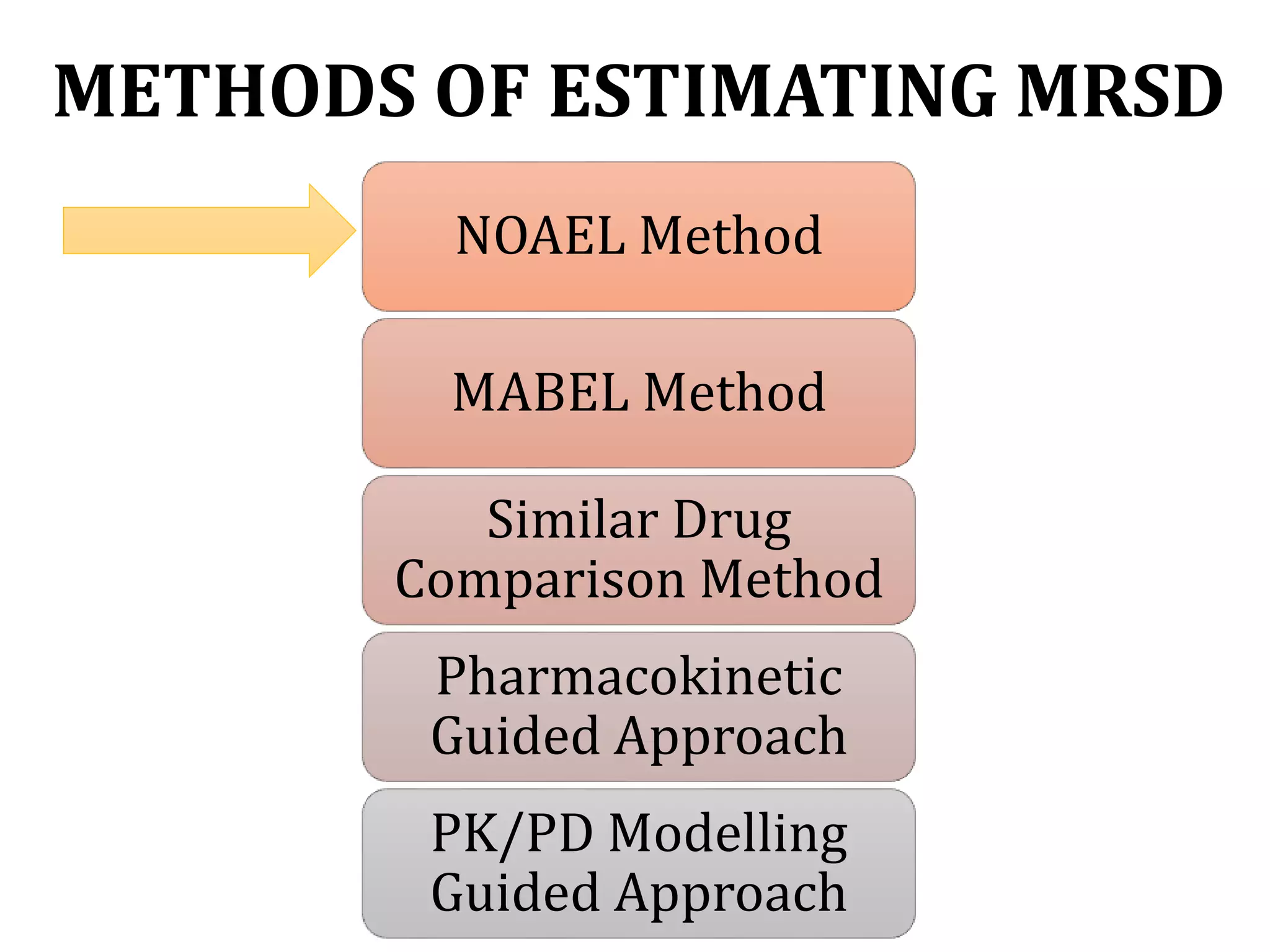

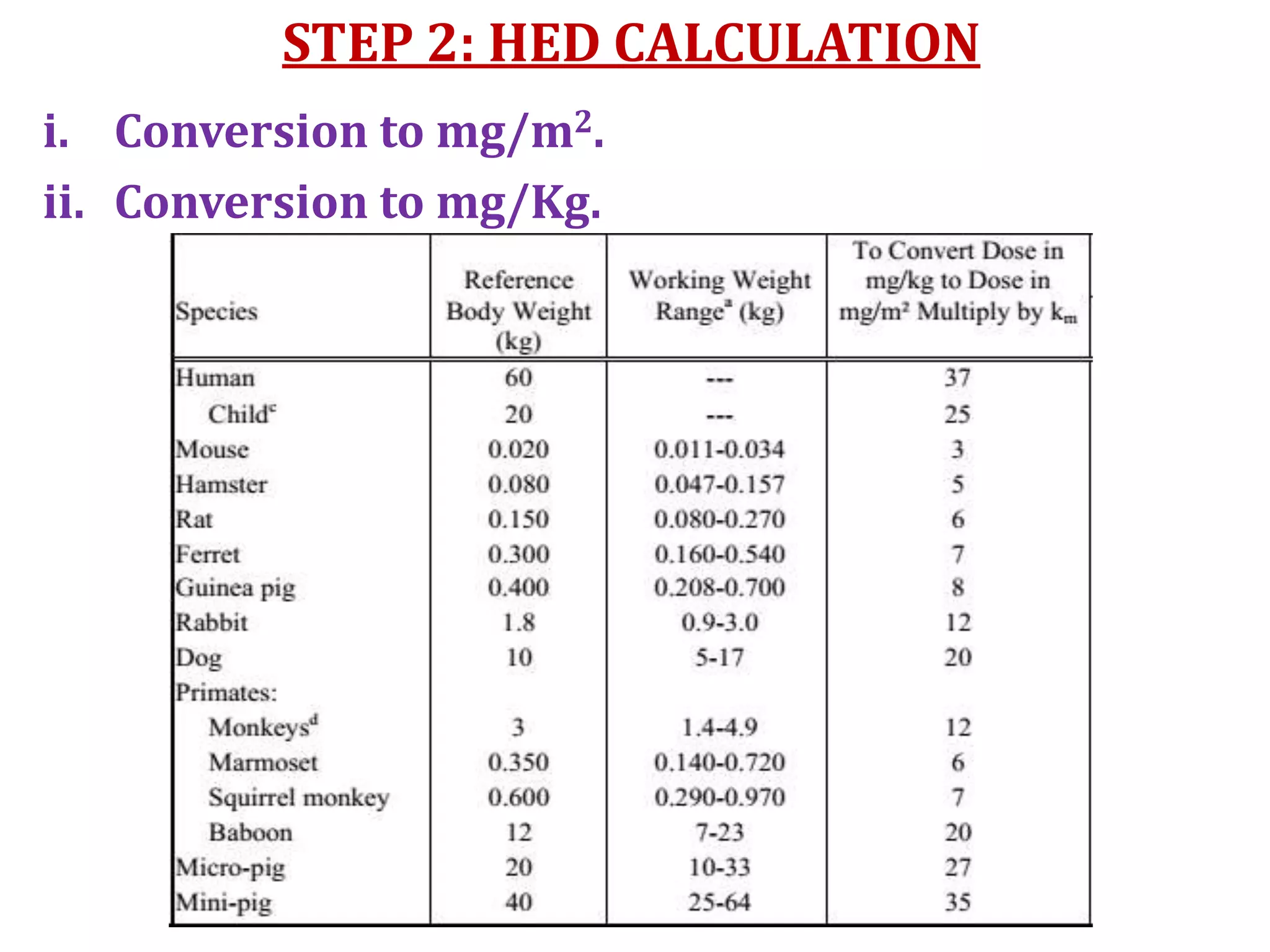





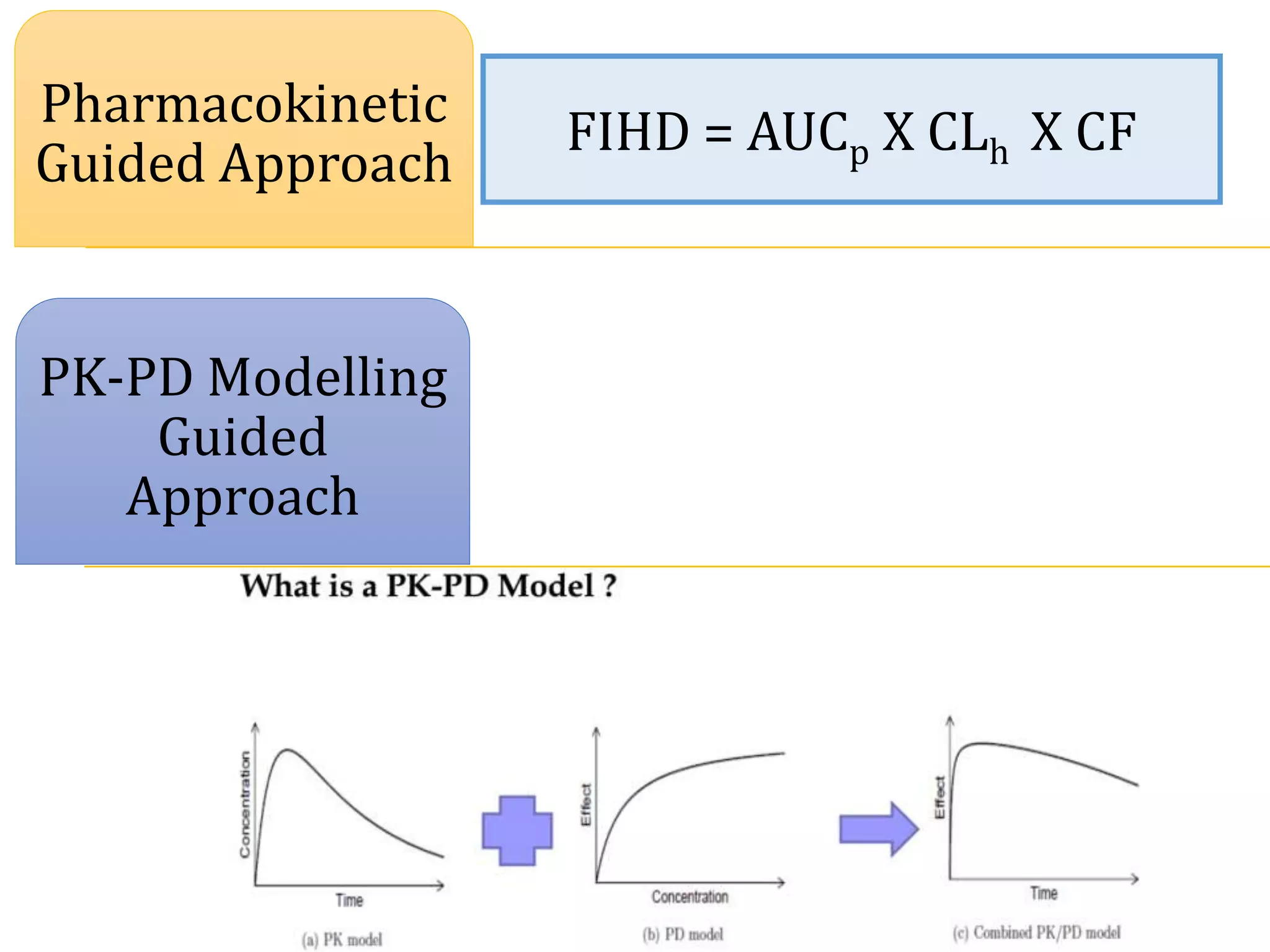










This document discusses approaches for determining appropriate doses in preclinical and clinical studies. It covers considerations for in vitro, in vivo animal, and first-in-human clinical doses. For animal studies, a maximum tolerated dose is determined through dose range finding studies and acute toxicity studies. Regulatory toxicology studies use doses including a low dose at the no-observed-adverse-effect level, intermediate doses, and a high dose close to the maximum tolerated dose. For first-in-human studies, the estimated dose is typically 1/10 of the human equivalent dose calculated from the no-observed-adverse-effect level in the most appropriate animal species. Pharmacokinetic modeling and other drug properties may further inform safe starting doses























































































