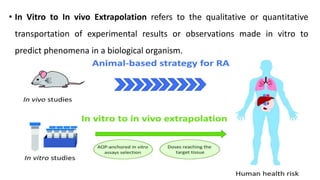
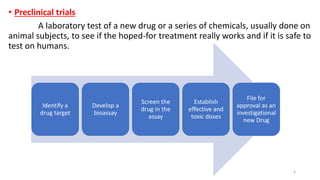
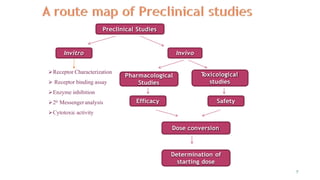
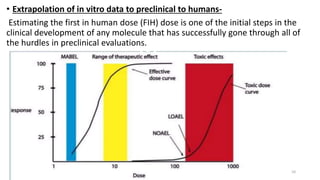
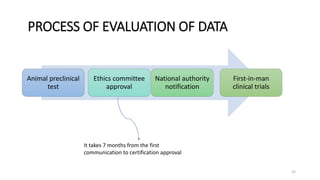
This document discusses extrapolating data from in vitro studies to preclinical and human trials. It defines extrapolation as estimating conclusions based on known facts. Two main methods of extrapolation are described: linear scaling and allometric scaling. When estimating a first human dose, the no-observed adverse effect level from animal studies is determined and converted to a human equivalent dose using body surface area. A safety factor is then applied to determine the maximum recommended starting dose. The document also discusses other approaches like using the minimum anticipated biological effect level.