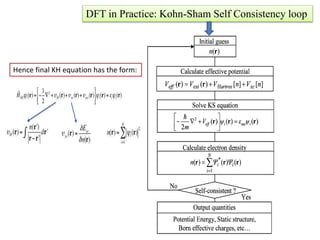
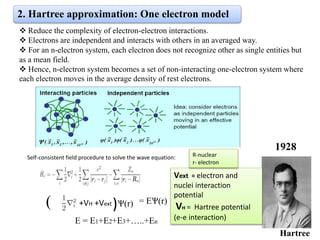
The document discusses Density Functional Theory (DFT) and its foundational concepts, including the Schrödinger equation, the Born-Oppenheimer approximation, and various methods for addressing many-body problems in quantum mechanics. It highlights the development of DFT as a means to simplify complex calculations by focusing on electron density instead of wave functions, while detailing various computational codes like VASP and SIESTA. Key historical contributions from scientists such as Hohenberg, Kohn, and Sham are emphasized, alongside the introduction of approximation techniques like the Local Density Approximation (LDA) and Generalized Gradient Approximation (GGA).








![5. Kohn- Sham Approach (1965):
KS replace the interacting n-electron system with a system of one-electron (non-
interacting) system in effective potential having the same ground state.
since the kinetic energy; E= Ekin+ Eext+EH +Ex+ Ec
int
non
non int
Ekin = Ekin + Ekin
where
E = Ekin + Ekin + Eext + EH +Ex + Ec
int
non int
E = Ekin + Eext + EH +Exc = F [n(r)] + Eext
non](https://image.slidesharecdn.com/basicsofdensityfunctionaltheory-240520051507-479d0037/85/Basics-of-Density-functional-theory-pptx-10-320.jpg)