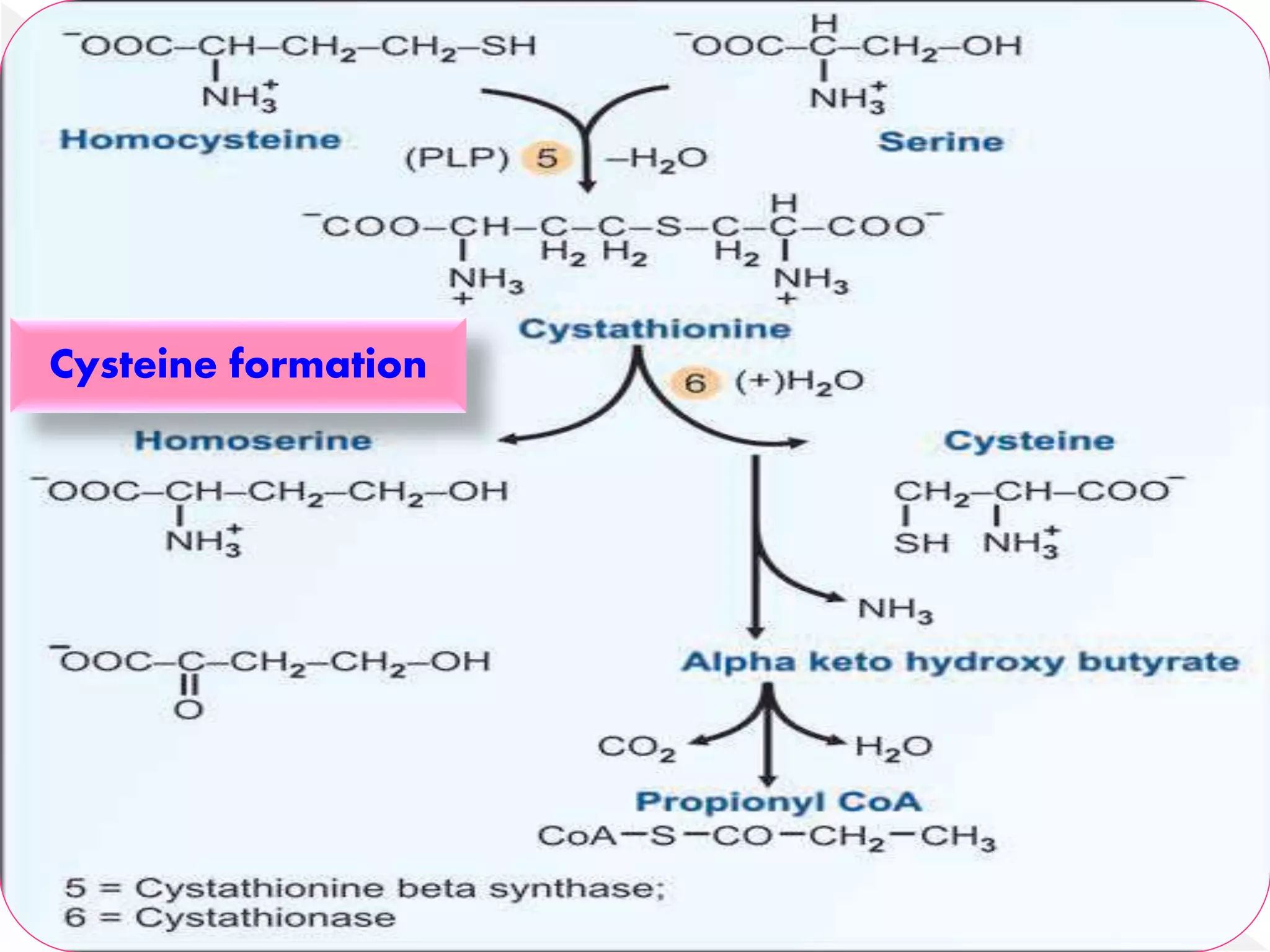
Cysteine is formed from methionine through a series of reactions involving homocysteine and serine. Homocysteine condenses with serine to form cystathionine, which is then cleaved by cystathioninase to form cysteine and α-ketobutyrate. Cysteine can be oxidized to form taurine or participate in glutathione synthesis. Defects in cysteine metabolism can cause cystinuria, cystinosis, and homocystinurias, characterized by accumulation of cystine, cysteine, or homocysteine respectively, potentially leading to organ damage.
It isnon-essential & glucogenic amino acid.
Cysteine is present in large quantity in
keratin of hair & nails.
Formation of cysteine is by using the carbon
skeleton contributed by serine & sulfur
originating from methionine.
3.
Homocysteine formedfrom methionine is a
precursor for the synthesis of cysteine.
Homocysteine condenses with serine to form
cystathionine.
4.
This reactionis catalysed by a PLP-
dependent cystathionine synthase.
The enzyme cystathioninase (PLP-dependent)
cleaves & deaminates cystathionine to
cysteine & α-ketobutyrate.
Homocysteine isan intermediate in the synthesis of
cysteine from methionine.
Elevation in plasma homocysteine (normal <15 pmol/l)
has been implicated in coronary artery disease.
Homocysteine reacts with collagen to produce
reactive free radicals, besides interfering with
collagen cross links.
Homocysteine is also involved in the aggregation of
LDL particle & leads to atherosclerosis.
8.
Cystine &cysteine are interconvertible by an
NAD+ dependent cystine reductase.
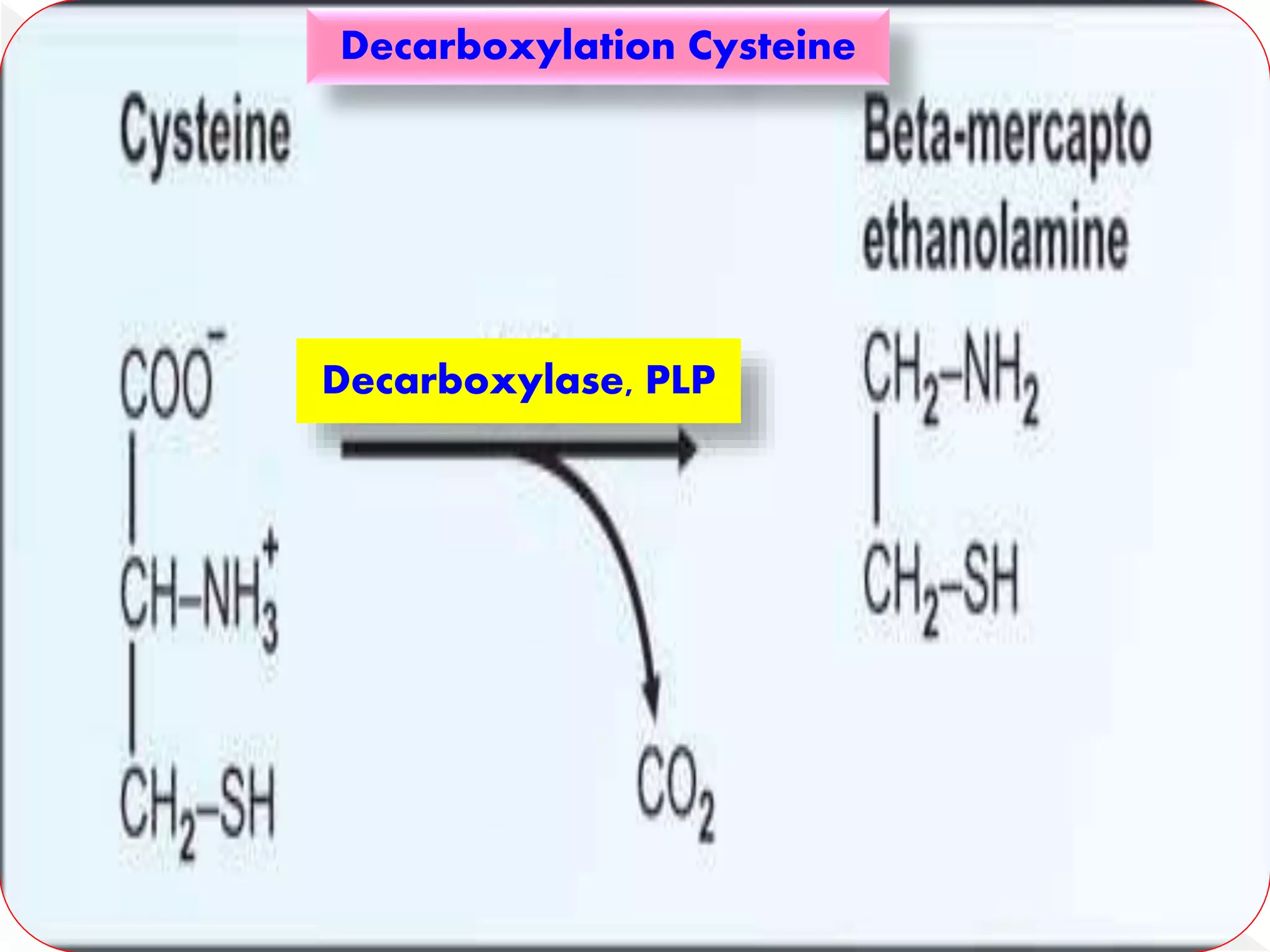
Cysteine on decarboxylation produces
mercaptoethanolamine which is involved in
the biosynthesis of coenzyme A from the
vitamin pantothenic acid.
The enzymecysteine dioxygenase oxidizes
cysteine to cysteine sulfinate, on further
oxidation, is converted to cysteic acid.
Cysteic acid undergoes decarboxylation to
produce taurine which conjugates with bile
acids.
Cysteic acid can also be degraded to
pyruvate, which is glycogenic.
Cysteine sulfinatecleaves off alanine to
produce sulfite which is converted to sulfate
& excreted in urine.
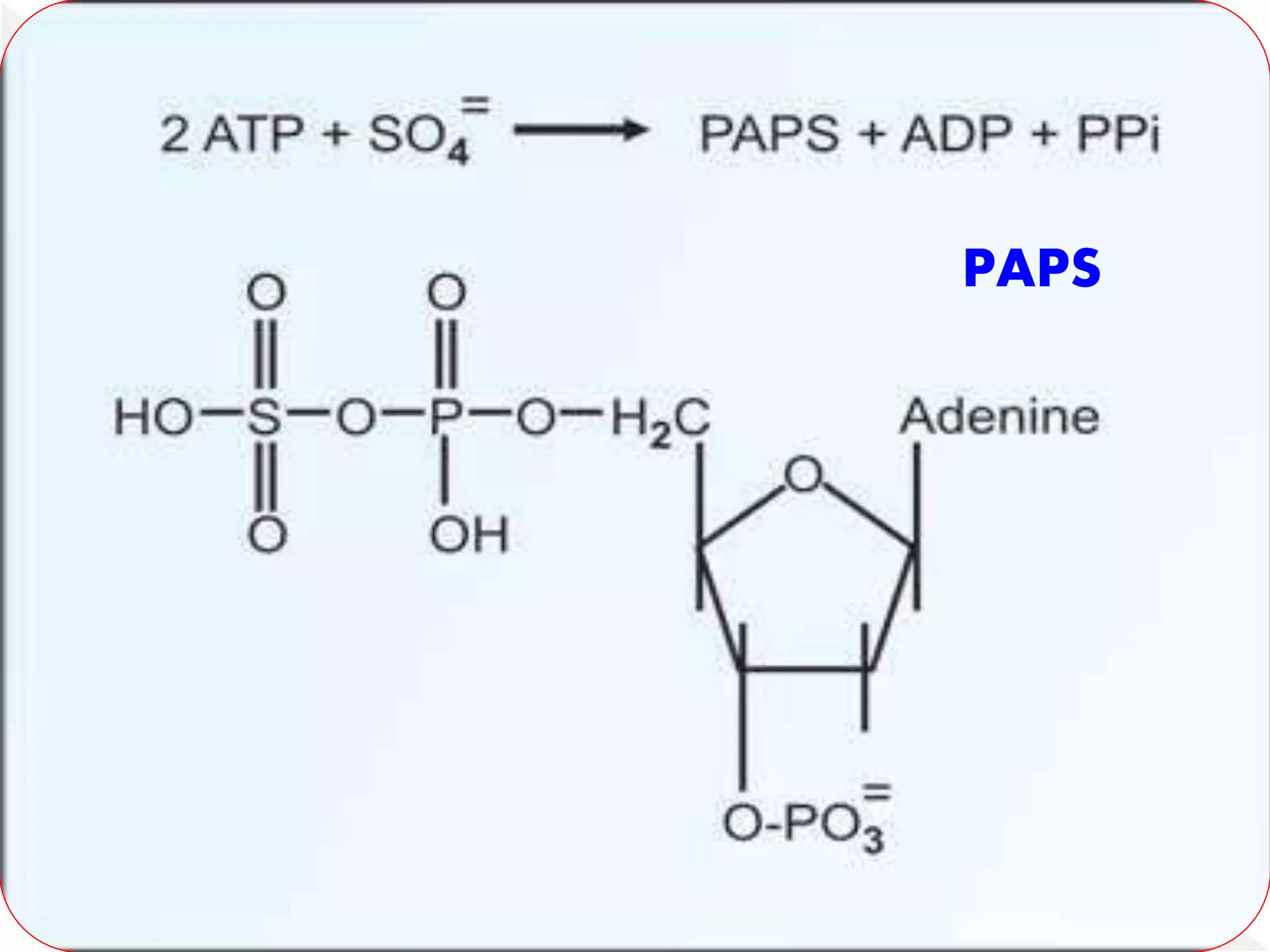
Some amount of sulfate condenses with ATP
to form active sulfate or 3'-
phosphoadenosine 5'-phosphosulfafe (PAPS).
13.
Active sulfate(PAPS) is utilized for the
synthesis of mucopolysaccharides.
Used in detoxification.
Sulfate is also a structural component of
proteins & lipids.
Cysteine can be degraded by desulfhydrase to
liberate sulfur (as H2S), ammonia & pyruvate.
Cysteine is a component of glutathione.
Cysteine isrequired for formation of
Glutathione.
Glutathionine is required for transport of
amino acids.
Glutathione is present in the RBCs.
This is used for inactivation of free radicals
formed inside RBC.
16.
Glutathione helpsto detoxify several
compounds.
Glutathione keeps the enzymes in reduced,
active state.
Cysteine residues in polypeptide chains form
disulfide bridges to make active proteins,
e.g. insulin & immunoglobulins.
17.
Cystinuria (cystine-lysinuria):
The most common inherited disease.
It is characterized by increased excretion of
cystine (25-40 times normal).
Elevation in the urinary output of lysine,
arginine & ornithine is also observed.
18.
A specificcarrier system exists in kidney
tubules for the reabsorption of amino acids,
namely cysteine, ornithine, arginine & lysine
(COAL to recall).
In cystinuria, this carrier system becomes
defective leading to the excretion of all these
four amino acids in urine.
19.
Cystine isrelatively insoluble & increased
concentrations leads to precipitation &
formation of cystine stones in kidney & urinary
tract.
Cystinuria is usually identified in the
laboratory by cyanide nitroprusside test.
The treatment includes restricted ingestion of
dietary cystine & high intake of fluids.
20.
Cystine crystalsare deposited in many tissues
& organs of reticuloendothelial system
throughout the body.
These include spleen, lymph nodes, liver,
kidney, bone marrow etc.
A defect in the Iysosomal function.
Cystine accumulates in the lysosomes of
various tissues.
21.
In cystinosis,renal function is impaired.
It is characterized by generalized amino
aciduria.
The affected patients die within 10 years,
mostly due to renal failure.
It is also due to defect in the enzyme cystine
reductase (interconverting enzyme).
22.
Homocystinurias area group of metabolic
disorders characterized by the accumulation
& increased urinary excretion of
homocysteine & S-adenosylmethionine (SAM).
Plasma concentration of methionine is
increased.
23.
Enzyme defect:Cystathionine synthase.
Accumulation of homocystetne.
It results in various complications-thrombosis,
osteoporosis & mental retardation.
The deficiency of cystathionine is associated
with damage to endothelial cells which might
lead to atherosclerosis.
24.
Two formsof type I homocystinurias
One of them can be corrected with vitamin B6
supplementation (B6 responsive) while the
other does not respond to B6.
The treatment includes consumption of diet
low in methionine & high in cystine.
The patients of homocystinuria have high
levels of homocysteine & usually die of
myocardial infarction, stroke.
25.
Homocystinuria ll:
N5 - N10 - Methylene THF reductase.
Homocystinuria lll:
N5 - N10 - Methyl THF homocysteine methyltransferase.
This is mostly due to impairment in the synthesis of
methylcobalamin.
Homocystinurla lV:
N5 - Methyl THF homocysteine methyl transferase.
This is primarily due to a defect in the intestinal
absorption of vitamin B12.
26.
Textbook ofBiochemistry - U Satyanarayana
Textbook of Biochemistry - DM Vasudevan






































































![Amino-Acid Metabolism [Methionine].pptx..](https://cdn.slidesharecdn.com/ss_thumbnails/amino-acidmetabolismmethionine-241015153732-4ddd3f27-thumbnail.jpg?width=640&height=640&fit=bounds)
























































