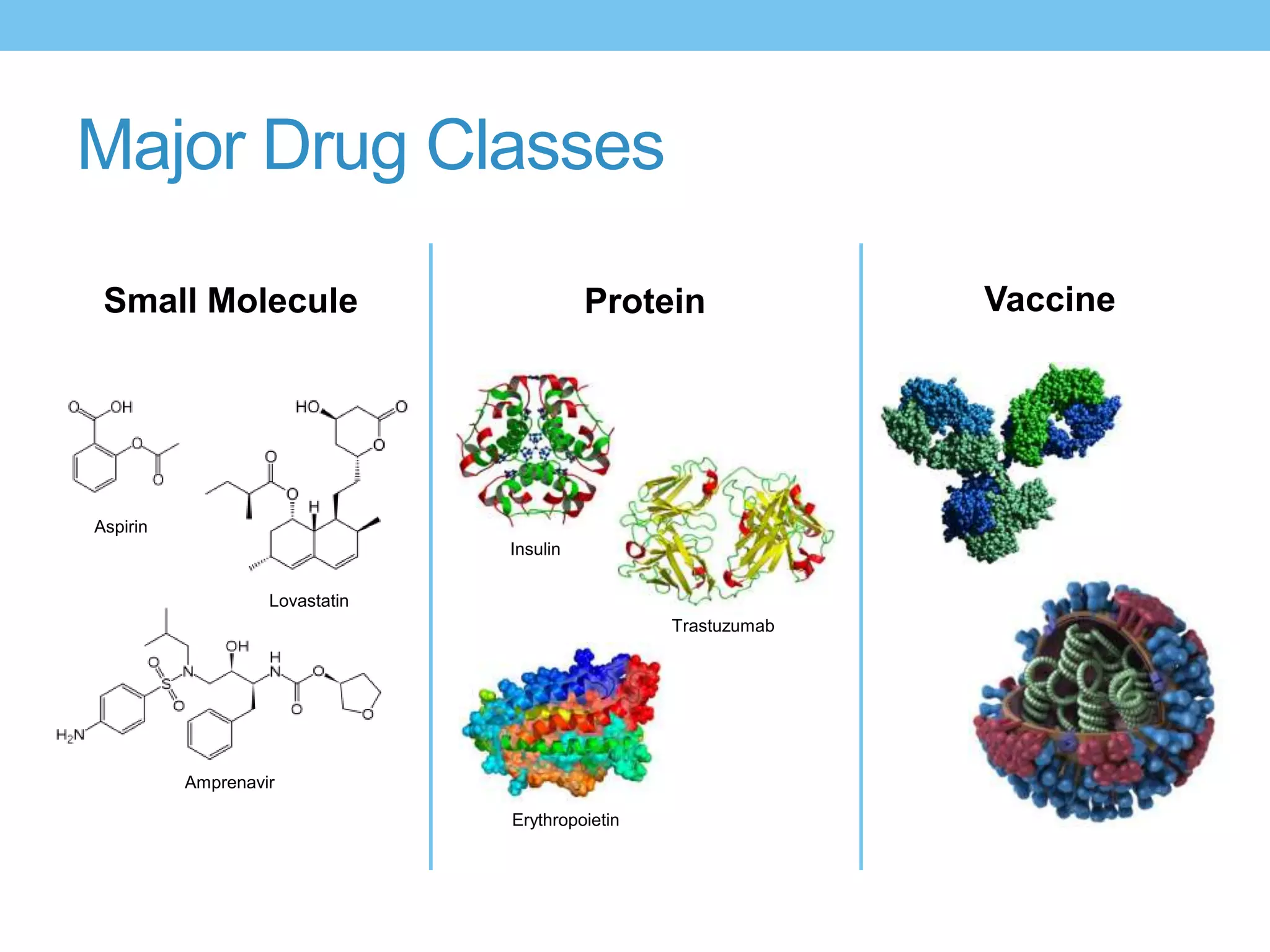
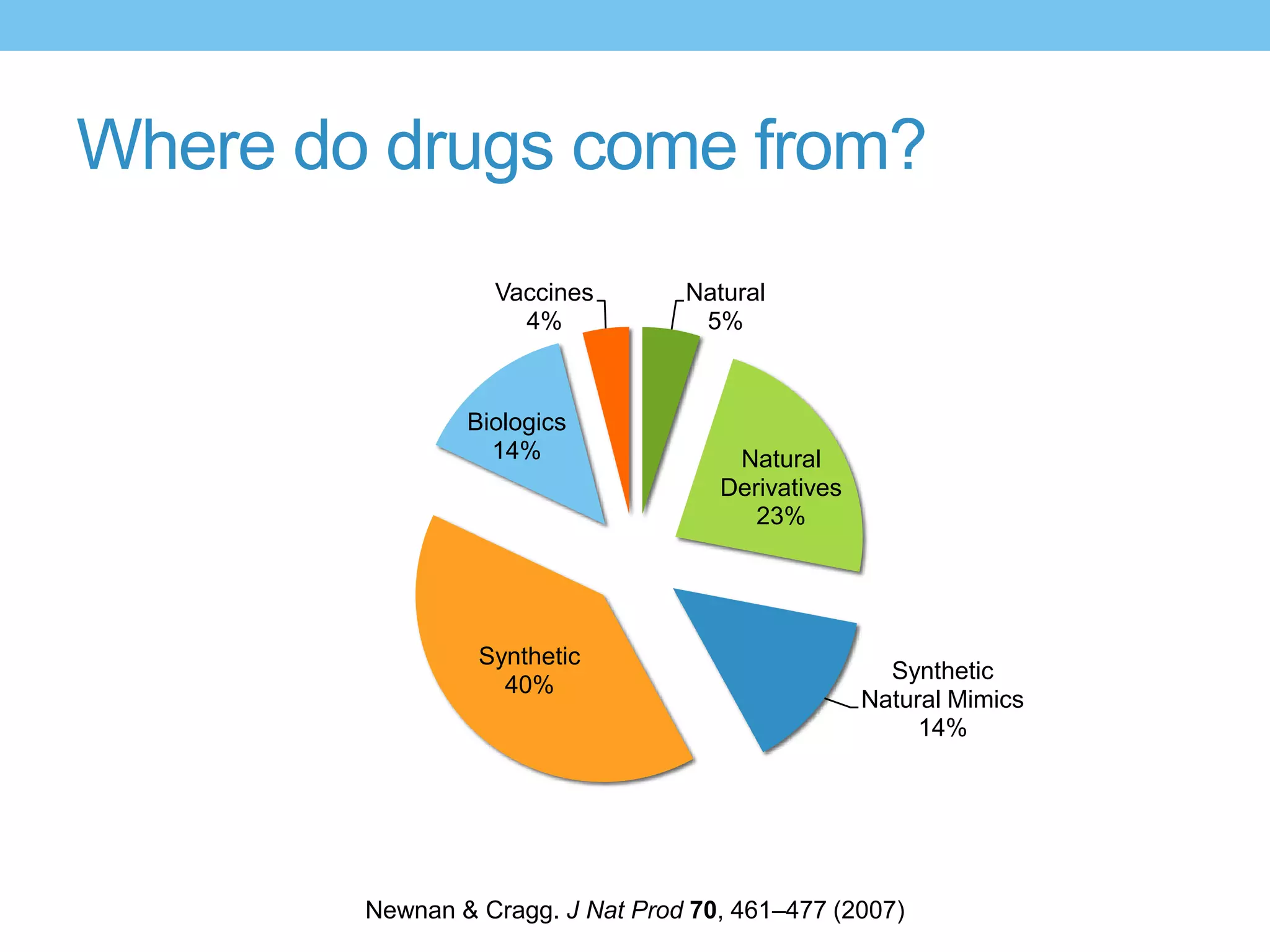
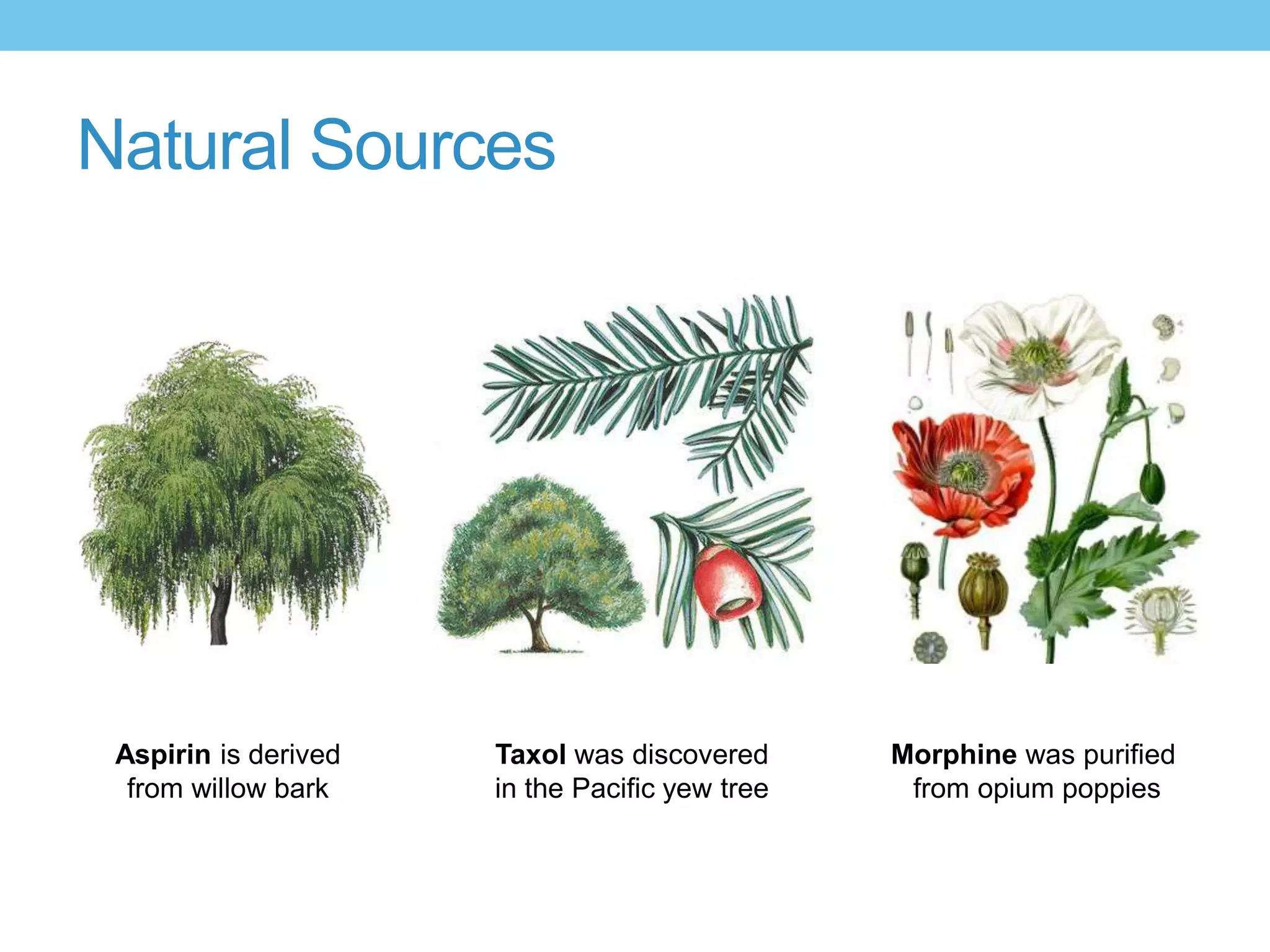
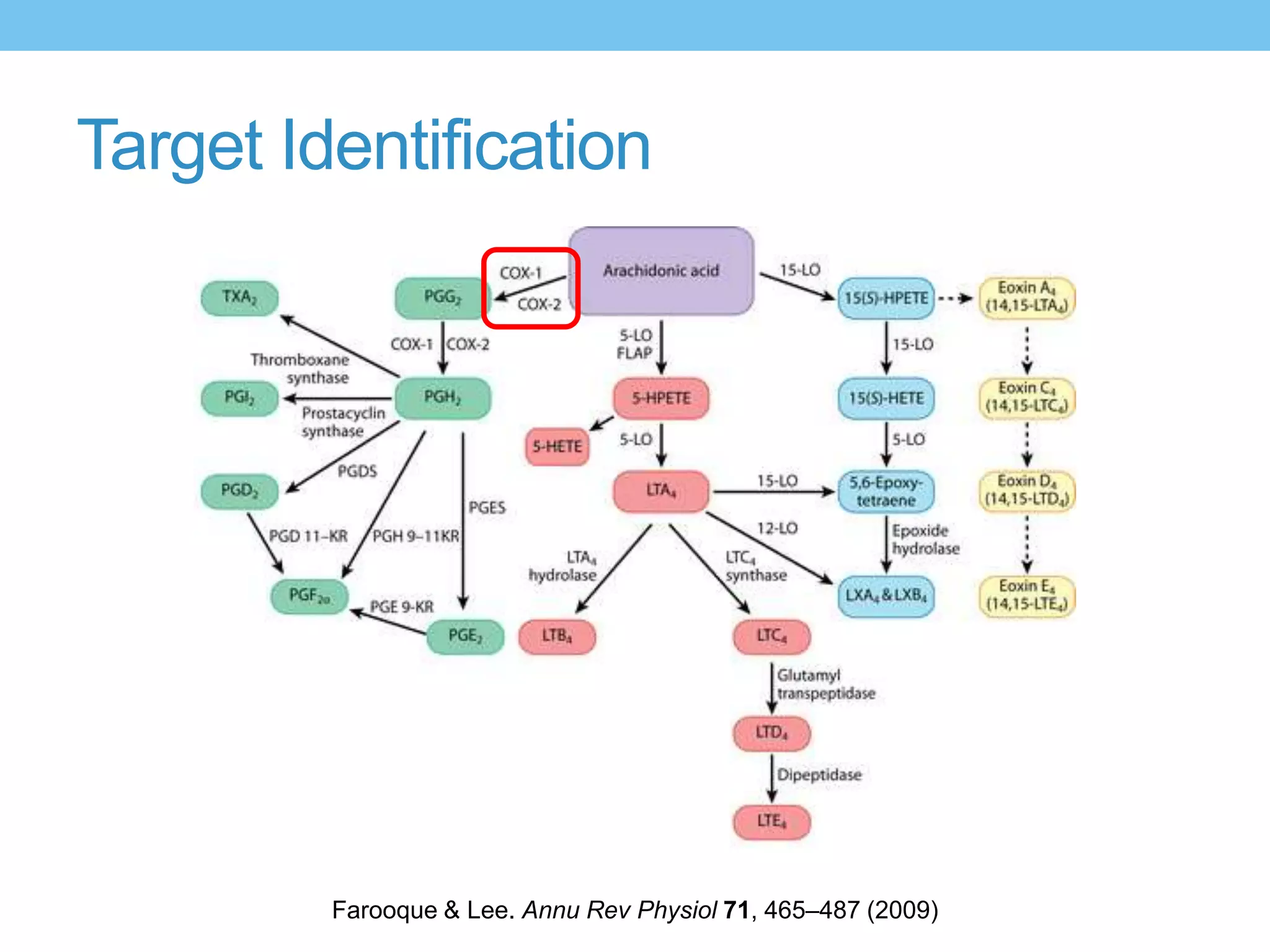
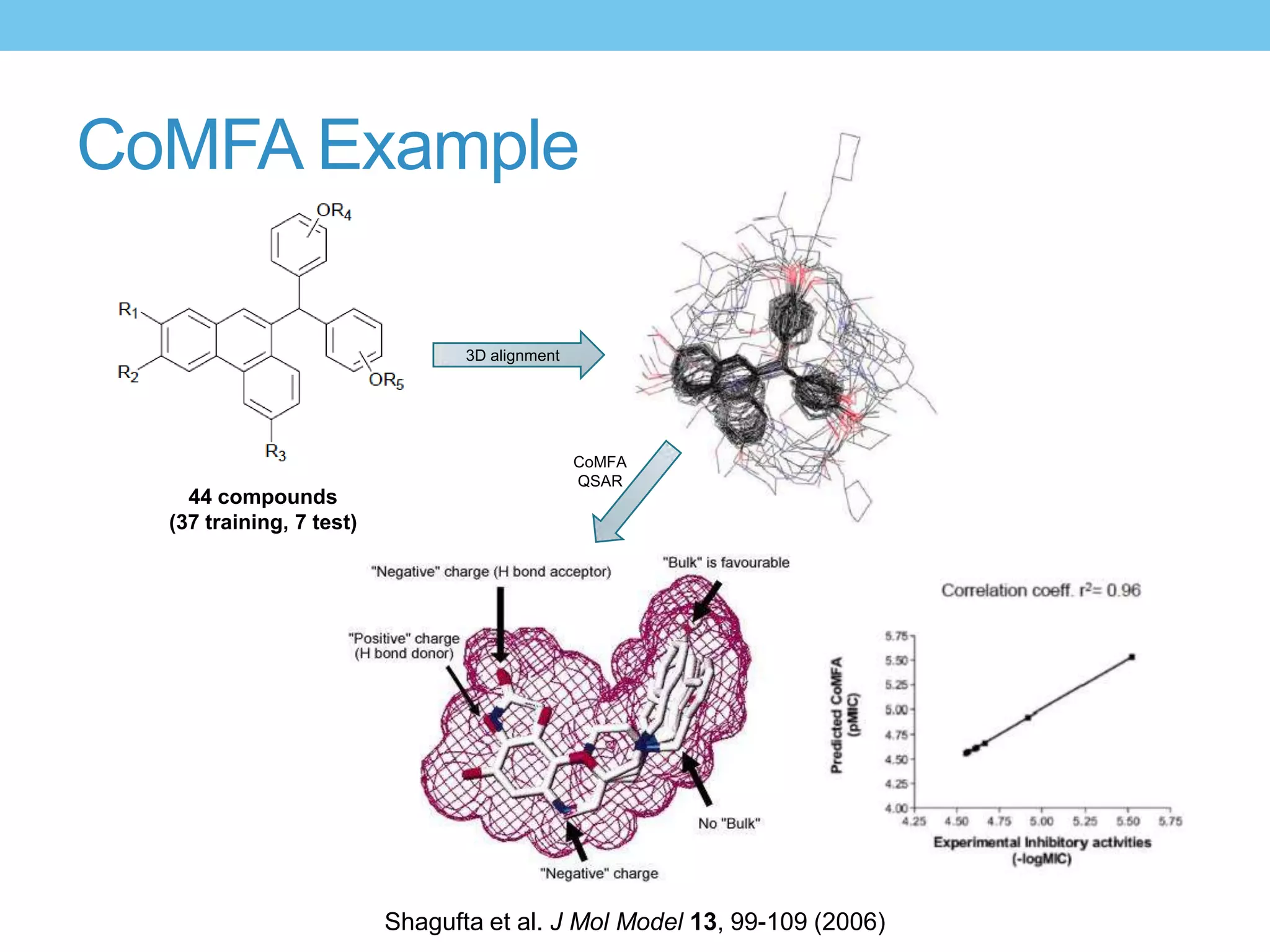
The document provides an overview of computer-aided drug design, outlining the pharmaceutical pipeline and methodologies like structure-based and ligand-based drug design. It discusses how drugs are sourced, their classes, and the impact of computer-aided design on drug discovery, including techniques like molecular docking and QSAR (Quantitative Structure-Activity Relationship). Additionally, it highlights challenges in modeling, pharmacophore modeling, and the role of molecular descriptors in predicting biological activity.














![Knowledge-Based Scoring Functions
Example: ligand carboxyl O to protein histidine N
Procedure:
1. Find all PDB structures with ligand carboxyl O
2. Compute all distances to protein histidine N’s
3. Plot histogram of all O-N distances: p(rO-N)
4. Calculate E(r) using inverse Boltzmann
Boltzmann: p(r) ~ exp[ -E(r)/(RT) ]
Inverse Boltzmann: E(r) = -RT ln[ p(r) ]
Muegge & Martin. J Med Chem 42, 791-804 (1999)](https://image.slidesharecdn.com/caddlecture-121015214843-phpapp02/75/CADD-Lecture-24-2048.jpg)





































































![Rheumatic Fever CASE PRESENTATION [Autosaved].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/casepresentationautosaved-251123182512-9d9b0da4-thumbnail.jpg?width=640&height=640&fit=bounds)








