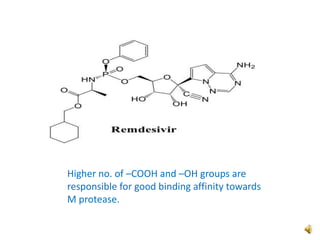
This document summarizes a presentation on tools and protocols for drug design using density functional theory (DFT). It introduces computer-aided drug design and molecular modeling techniques like quantum mechanics, semi-empirical methods, and DFT. Applications of these methods include structure optimization, calculating properties like HOMO-LUMO energies, and molecular docking for drug discovery. Several examples are provided of using DFT calculations to model drug-receptor binding and evaluate compounds for treating diseases.