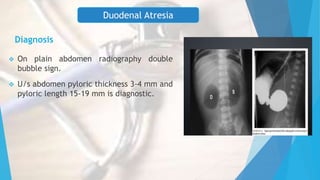
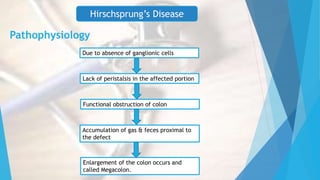
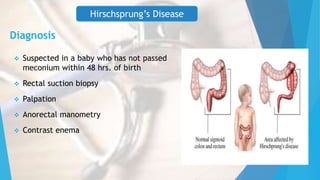
This document discusses common surgical emergencies in pediatrics, including tracheoesophageal fistula, duodenal atresia, Meckel’s diverticulum, Hirschsprung’s disease, appendicitis, and biliary atresia. Each condition is detailed with its definition, clinical features, diagnostic methods, treatment options, and complications. Key points and clinical questions are also highlighted for educational purposes.