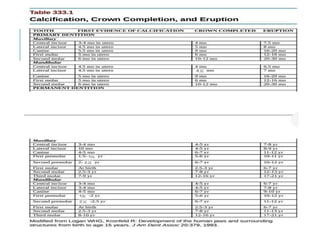
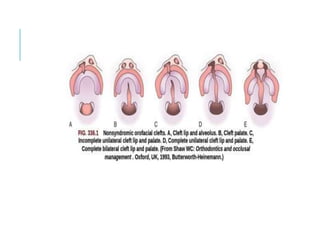
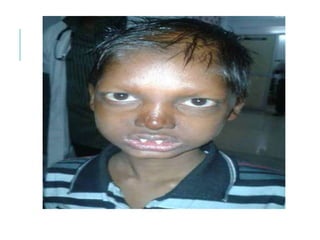
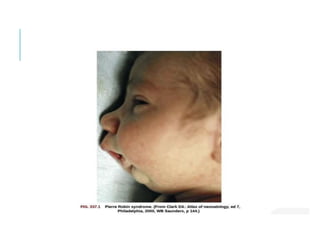
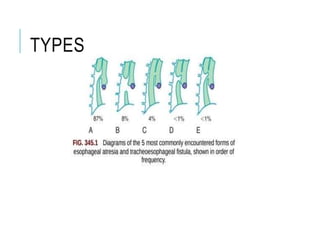
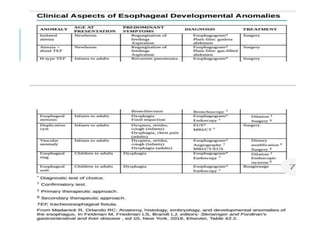
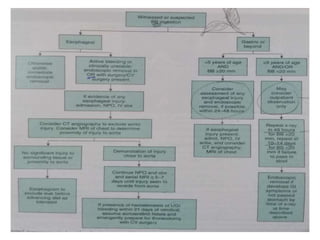
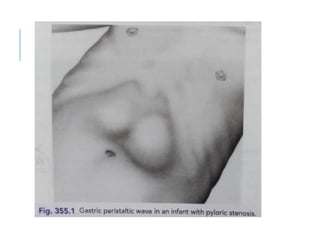
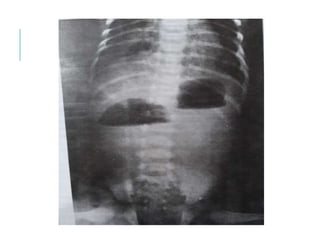
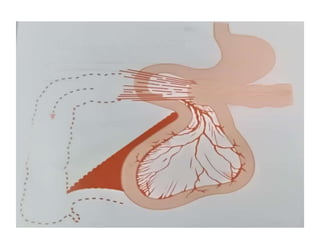
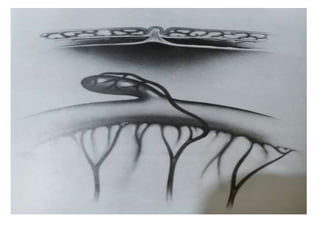
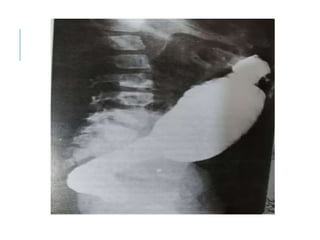
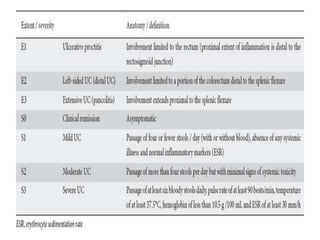
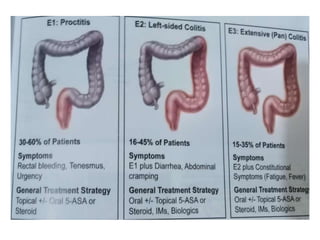
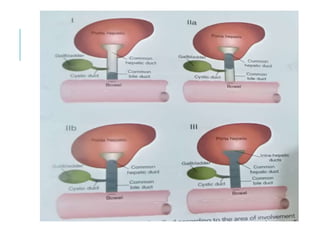
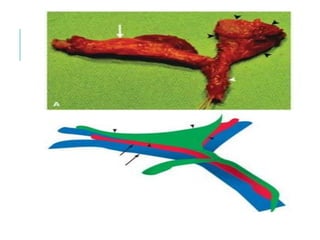
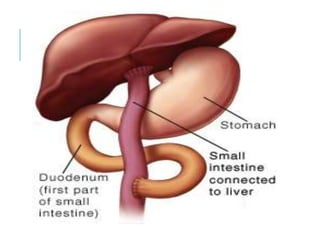
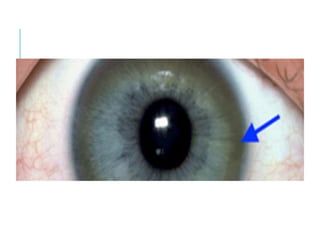
The document outlines various pediatric digestive system diseases, beginning with fetal swallowing processes and infant feeding preferences. It discusses anatomical variations, conditions like cleft lip and palate, and diagnoses and treatments for issues such as gastroesophageal reflux disease, foreign body ingestion, pyloric stenosis, and malrotation. The text emphasizes a multidisciplinary approach to treatment and the importance of timely surgical and medical interventions.