Downloaded 63 times






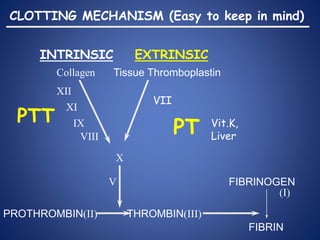













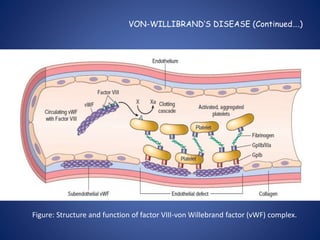





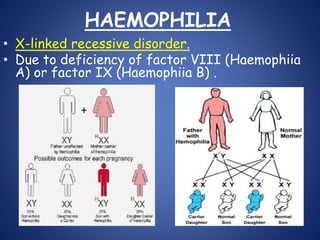


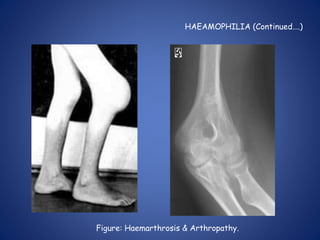




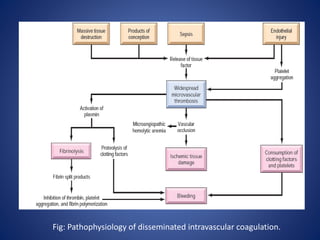







This document provides an overview of coagulopathy in children. It begins with objectives and definitions. It then discusses various types of bleeding disorders including quantitative and qualitative platelet disorders, coagulation factor disorders like hemophilia, and acquired disorders like disseminated intravascular coagulation. For each condition, it covers causes, clinical features, laboratory findings, and treatment strategies. It concludes with perioperative considerations and strategies for managing coagulopathy in children undergoing surgery.










































































