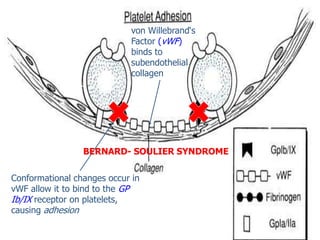
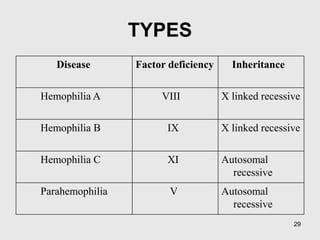
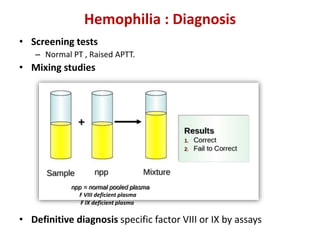
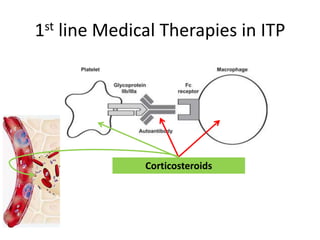
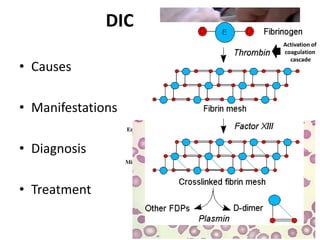
A 6-year-old boy presented with recurrent painful swelling of the left knee joint since age 2 and a history of prolonged bleeding from cuts. On examination, his left knee was swollen and tender. Laboratory tests showed a normal prothrombin time but elevated activated partial thromboplastin time that corrected with factor IX-deficient plasma, confirming a diagnosis of hemophilia B. The boy was advised to receive factor IX replacement therapy if he required a dental tooth extraction to prevent uncontrolled bleeding.