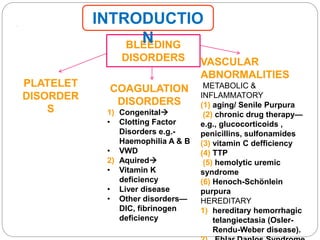
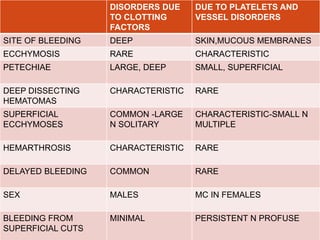
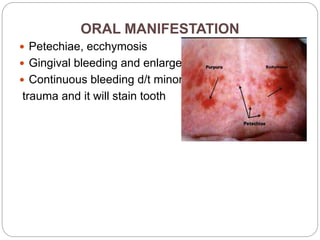
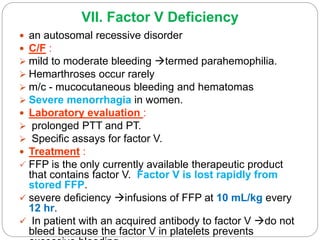
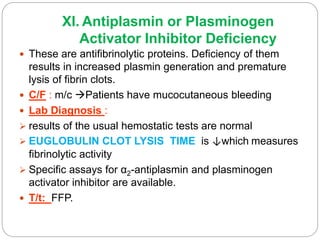
This document discusses bleeding disorders in children. It covers the main types which are platelet disorders, coagulation disorders, and vascular abnormalities. Coagulation disorders can be congenital like hemophilia A and B which are caused by clotting factor deficiencies, or acquired like those caused by vitamin K deficiency or liver disease. Hemophilia A is caused by a factor VIII deficiency while hemophilia B is a factor IX deficiency. Clinical manifestations of hemophilia include easy bruising, joint bleeding, and bleeding from minor injuries that persists. Treatment involves replacing the missing clotting factor through infusions to achieve hemostatic levels.































































![TREATMENT
The first 2 steps in the of DIC are the most critical:
(1) treat the trigger that caused DIC
(2) restore normal homeostasis by correcting the shock,
acidosis, and hypoxia that usually complicate DIC.
Replacement Therapy
•platelet infusions (for
thrombocytopenia)
•cryoprecipitate (for
hypofibrinogenemia)
• fresh frozen plasma (for
replacement of other
coagulation factors and
natural inhibitors).
In DIC associated with sepsis,
a controlled trial of
drotrecogin-α (activated
protein C concentrate [APC])
in adults is given. The role of
these agents in childhood
remains to be defined.
The role of heparin in DIC is
limited to patients who have
vascular thrombosis in
association with DIC.](https://image.slidesharecdn.com/bleedingdisordersinchildren-180102143754/85/Bleeding-disorders-in-children-69-320.jpg)







![DIFFERENTIAL DIAGNOSIS
medication that induces drug-dependent antibodies
splenic sequestration due to previously unappreciated
portal hypertension
aplastic /Fanconi anemia .
NONIMMUNE causes hemolytic-uremic syndrome
[HUS], disseminated intravascular coagulation [DIC]
hypersplenism owing to either liver disease or portal
vein thrombosis.
Autoimmune thrombocytopenia with insidious onset may
be an initial manifestation of SLE or rarely lymphoma.
Wiskott-Aldrich syndrome must be considered in
young males found to have low platelet counts,
particularly if there is a history of eczema and recurrent
infection.](https://image.slidesharecdn.com/bleedingdisordersinchildren-180102143754/85/Bleeding-disorders-in-children-77-320.jpg)




































































































