Downloaded 483 times









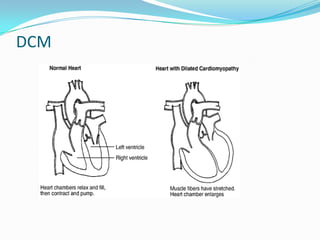















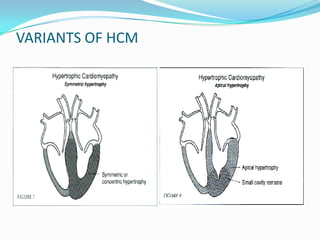
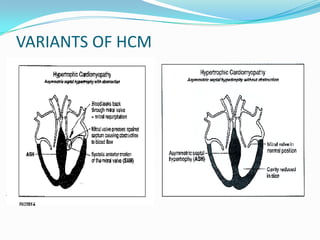
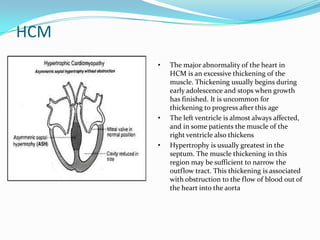
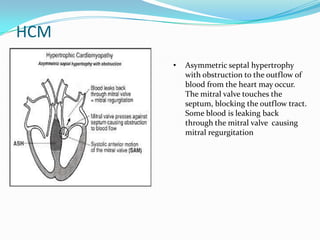


























This document discusses different types of cardiomyopathies including their classification, etiology, and treatment. It focuses on dilated cardiomyopathy (DCM), providing details on its classification, causes, clinical presentation, diagnosis, and management. It also discusses other forms of cardiomyopathy such as hypertrophic cardiomyopathy (HCM), describing its characteristic left ventricular hypertrophy and outflow tract obstruction. The document aims to comprehensively classify and describe different cardiomyopathies for medical professionals.























































































