Download as PDF, PPTX







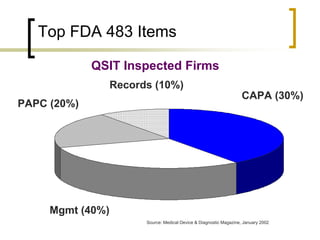























A CAPA (corrective and preventative action) program is an important indicator of a company's overall compliance efforts. It is considered a "bellwether" by the FDA. An effective CAPA program follows a closed-loop process to identify, correct, and eliminate quality issues and potential problems. It analyzes multiple sources of quality data. Failing to properly establish and maintain CAPA procedures is a common violation cited by the FDA. Ensuring a strong CAPA program that satisfies regulations like ISO 13485 and 21 CFR Part 820 can help reduce compliance risks during FDA inspections.















































