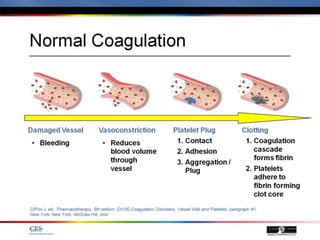
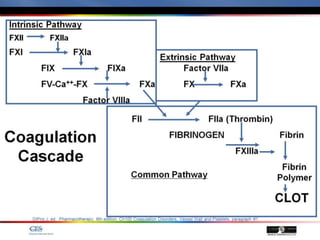
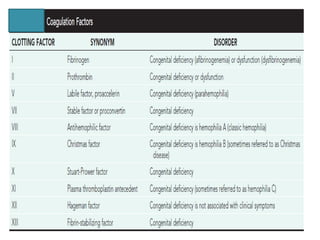
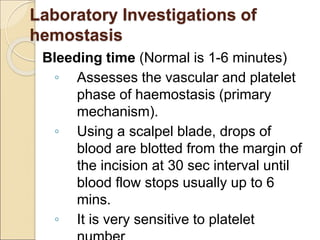
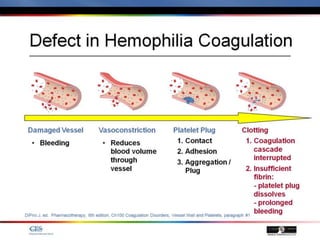
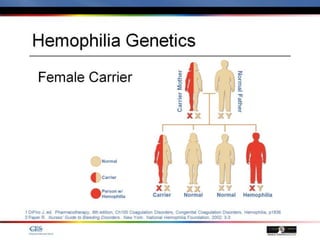
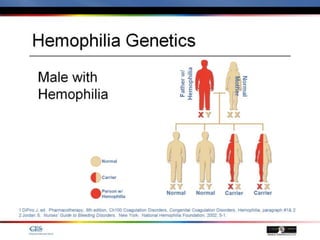
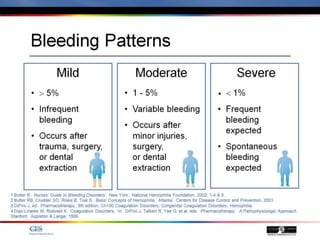
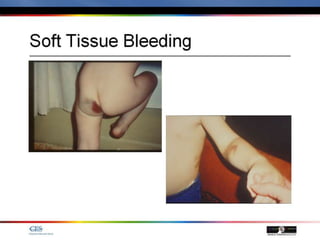
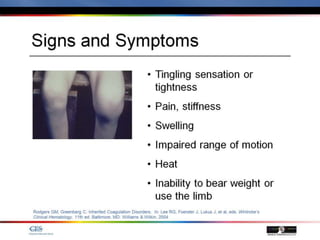
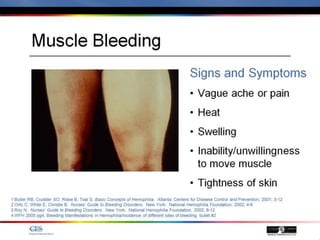
The document discusses idiopathic (autoimmune) thrombocytopenic purpura (ITP). ITP is the most common cause of acute thrombocytopenia in otherwise healthy children. It typically presents as a previously healthy 1-4 year old with sudden onset of petechiae and purpura, often following a viral illness. Laboratory findings show severe thrombocytopenia and a normal bone marrow with increased megakaryocytes. The condition is usually self-limiting and resolves within 6 months in 80% of children.