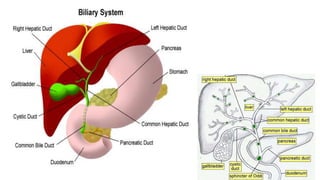
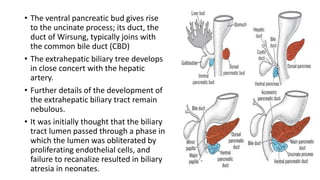
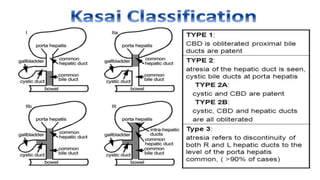
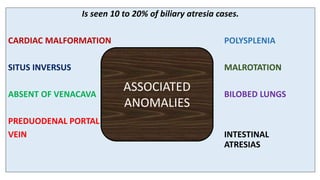
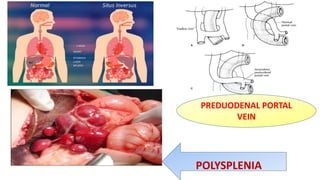
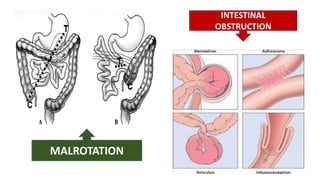
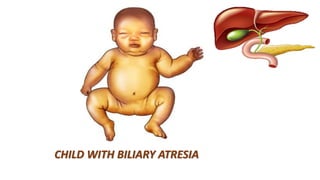
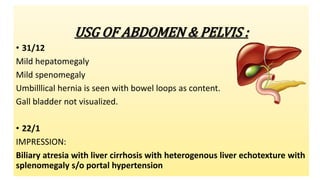
1) Biliary atresia is a condition where the bile ducts inside and outside the liver are scarred and blocked, preventing normal bile flow from the liver to the small intestine.
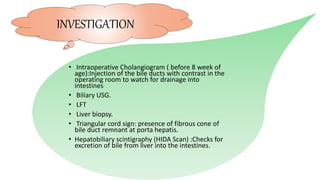
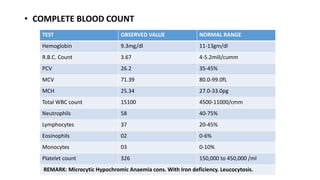
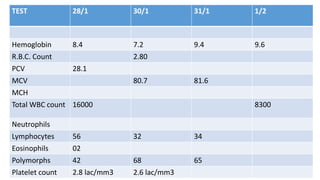
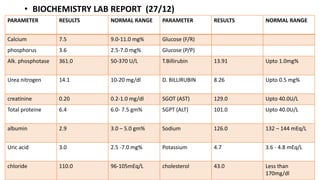
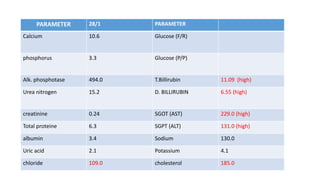
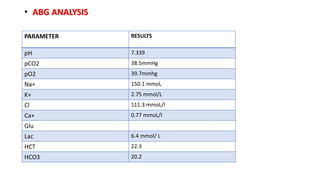
2) The patient, an infant, presented with prolonged jaundice, dark urine, light-colored stools and was found to have biliary atresia. Investigations including blood tests and imaging confirmed the diagnosis.
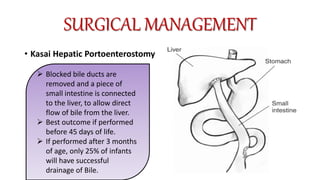
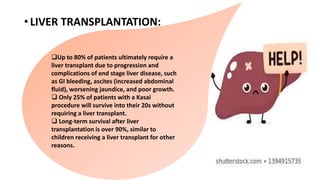
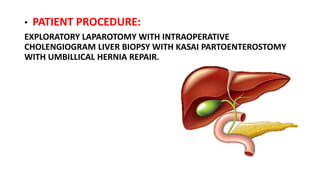
3) The patient underwent a Kasai procedure to connect the liver directly to the intestine to restore bile flow, along with hernia repair. Long term complications include liver failure requiring transplantation in 80% of patients.