INTRODUCTION
• Encephalitis isan inflammatory condition of brain with many etiologies
• Autoimmune encephalitis: Group of clinical syndromes with an autoimmune pathogenic basis,
associated with antibodies against neuronal cell surface proteins and synaptic receptors involved in
synaptic transmission, plasticity or neuronal excitability
• The syndromes vary according to associated antibody
• Most of these disorders are severe and potentially fatal, but frequently respond to immunotherapy with
good outcomes
PATHOGENESIS
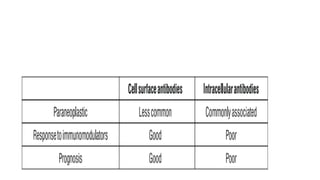
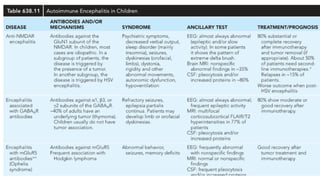
● AIE categorizedas per antigen location into 2 groups
● Antibodies that target intracellular antigens and that target cell surface antigens
CELL SURFACE AB INTRACELLULAR AB
Anti NMDAR(MC) Anti-Hu
LGl1 Anti-Ma2
VGKC Anti-GAD
CASPR2
Anti-GABA-A
Anti-GABA-B
MOG
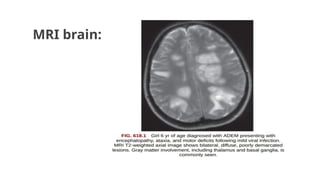
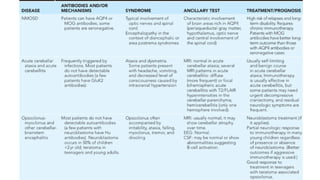
ADEM:
◦ MC causeof autoimmune encephalitis in children and adolescents.
◦ Acute onset of polyfocal neurological deficit accompanied by encephalopathy
and changes compatible with demyelination on MRI brain.
12.
EPIDEMIOLOGY:
●It can occurat any age most cases reported 5-8 yrs, slight male predominance.
●Usually it is monophasic, if recurrence occur 3 month or longer after the first episode termed as
MDEM (Multiphasic Disseminated Encephalo-Myelitis).
●50% ADEM MOG (Myelin Oligodendrocyte Glycoprotein) Ab +, almost all case of MDEM MOG Ab +.
●ADEM can be followed by demyelination in a new location & if MOG-Ab negative - MS
●ADEM f/b relapse in specific location like
Optic nerve -ADEM-ON
● Optic nerve and spinal cord- NMOSD . Both frequently a/w MOG-Ab positive.
Diagnostic criteria forADEM
● A firstpolyfocal clinical CNS event with presumed inflammatory
demyelinating cause.
● Encephalopathy (unexplained by fever ,systemic illness or post
ictal symptoms)
● Brain MRI consistent with demylination during the acute (less
than 3 months ) phase
● No new clinical or MRI findings 3 months or more after the
clinical onset
15.
CLINICAL MANIFESTATION:
●Initial symptomsinclude- lethargy ,fever ,headache ,vomiting ,meningeal sign and seizure.
●Encephalopathy:
1. Is hallmark
2. Ranging from change in behaviour , persistent irritability to coma.
●Focal neurological deficit:
1. Difficult to ascertain in obtunded or very young child ,
2. Common neurological signs- visual loss, ataxia, motor and sensory deficit, bowel bladder dysfunction
if spinal cord demyelination occurs.
●It is usually rapidly progressive over days may need ICU admission particularly when
1. Brain stem dysfunction
2. Raised ICT.
16.
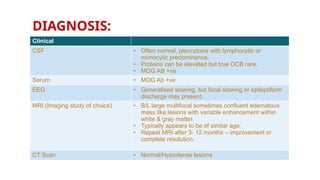
DIAGNOSIS:
Clinical
CSF • Oftennormal, pleocytosis with lymphocytic or
monocytic predominance.
• Proteins can be elevated but true OCB rare.
• MOG AB +ve
Serum • MOG Ab +ve
EEG • Generalised slowing, but focal slowing or epileptiform
discharge may present.
MRI (Imaging study of choice) • B/L large multifocal sometimes confluent edematous
mass like lesions with variable enhancement within
white & gray matter.
• Typically appears to be of similar age.
• Repeat MRI after 3- 12 months – improvement or
complete resolution.
CT Scan • Normal/Hypodense lesions
Treatment:
Symptomatic treatment
•Empirical antibioticsand antivirals should be considered when infective evaluations are pending
High dose steroids
•IV methylprednisolone- 20-30 mg/kg for 5 days (max. 1000mg/day)
•Followed by oral prednisolone 1-2 mg/kg/day over 4-6 weeks (max. 40-60 mg/day)
For refractory or severe cases
Others options
•IVIG – 2 gm/kg over 2-5 days
•Plasmapheresis- 5-7 exchange every other days
19.
Prognosis:
●Most children experiencesfull motor recovery.
●But residual defect may be seen- cognitive deficit & behavioural changes.
Differential diagnosis:
●Multiple sclerosis
●Leukodystrophy
●Vasculitis
●Tumor
●Ab associated disorder
21.
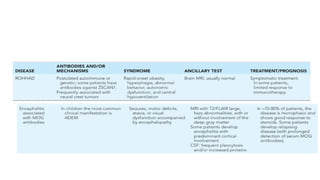
Anti NMDA ReceptorEncephalitis
• Anti NMDAR encephalitis is the 2nd
MCC of AIE after ADEM in children and adolescents
• Accounts for 4% of all encephalitis
• It is described as the prototype of AIE
• Incidence : 2.2/million children per year
• It also contribute to recurrence of encephalitis following HSV encephalitis in both children and
adults
22.
● Anti-NMDAR encephalitiswas 1st
described in 2007 as a paraneoplastic syndrome in adult females in
association with ovarian teratomas
● Since then it has been described in men, women and children of all age groups with and without
teratomas
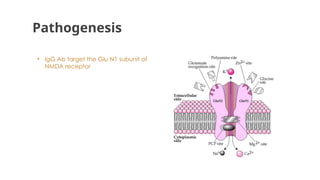
● Pathogenic IgG1 antibodies bind to GluN1 subunits of NMDA receptor leading to their
internalization- leads to inhibition of glutaminergic excitation to inhibitory neurons and in turn
intense excitotoxicity
TRIGGERS
● The mechanismsthat trigger the production of the antibodies are unknown in most cases
● PARANEOPLASTIC SYNDROMES:
• Results when tumor antigens are shared by neuronal cell antigens, leading to antibody mediated
immunological destruction of neuronal tissue
• Ovarian teratoma, testicular carcinoma, Hodgkin disease, neuroblastoma
25.
● POST VACCINE:
•Influenza, Polio, DPT, Japanese B encephalitis
● INFECTIONS:
• Mycoplasma pneumoniae, herpes simplex virus (HSV), human herpesvirus 6, enterovirus, COVID-19, and
influenza virus
• With the exception of HSV1, a pathogenic link with most of these infections has not been established
26.
● 50% ofpatients with HSV encephalitis develop antibodies against the GluN1 subunit of the
NMDAR and other neuronal cell surface proteins and receptors
● Half the patients develop new or relapsing neurologic symptoms 2-12 weeks after completing
treatment for HSV encephalitis
● In children younger than 4 years, It usually manifests with choreoathetosis and dyskinesias:
choreoathetosis post-HSV encephalitis
● A similar complication has been reported in patients with Japanese encephalitis
27.
Clinical features
● AntiNMDA-R encephalitis has been described in all ages, but more common in children and young
adults
● More common in females; but among <12 yrs, boys are more affected
● The resulting syndrome is highly predictable and usually evolves in stages
● Polysymptomatic syndrome with encephalopathy, movement disorders, psychiatric manifestations and
autonomic dysfunction
28.
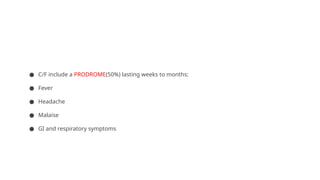
● C/F includea PRODROME(50%) lasting weeks to months:
● Fever
● Headache
● Malaise
● GI and respiratory symptoms
29.
● Prominent psychiatricmanifestations: In teenagers and young adults
● Rapidly progressive anxiety
● Agitation
● Delusions, Hallucinations
● Bizarre behavior, Labile affect, Mood disturbances (mania)
● Catatonic features
● Memory deficit
● Language disintegration
● Aggression
● Insomnia or other sleep disturbances
30.
● NEUROLOGICAL Manifestations
●Decreased level of consciousness
● Insomnia
● Mutism
● Seizures
● Cognitive deterioration
● Short term memory loss
● Central hypoventilation syndrome
● AUTONOMIC DYSFUNCTION
●Tachycardia/bradycardia
● Fluctuations in BP
● Arrhythmias
● Hyperthermia
● Sialorrhea
33.
● Toddlers andinfants: More frequently present with seizures and movement disorders
● Psychiatric-behavioral features may be missed
● Behavior changes include irritability, new-onset temper tantrums, agitation, aggression, reduced
speech, mutism, and autistic like regression.
● Some develop cerebellar ataxia and hemiparesis
● Autonomic dysfunction is usually milder and less severe in children
34.
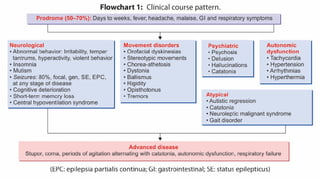
● ADVANCED DISEASE:Stupor, coma, periods of agitation alternating with catatonia as well as autonomic
dysfunction, respiratory failure
● Three clinical phenotypes: Classic form
Psychiatric form(good outcomes)
Catatonia predominant form(poor outcome)
36.
DIAGNOSIS
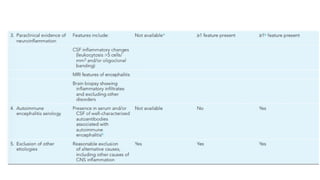
Features supportive ofAI etiology include:
● Evidence of CNS inflammation(CSF pleocytosis, elevated IgG index or oligoclonal bands,
elevated CSF neopterin)
● MRI abnormalities
● Response to immunosuppressive treatment
40.
CSF
● Initially abnormalin approximately 80% of patients
● CSF pleocytosis (lymphocytic)
● Normal/mild elevation in proteins
● Normal glucose
● Elevated IgG index
● Oligoclonal bands
● CSF neopterin
MRI BRAIN
● U/Lor B/L T2/FLAIR hyperintensities involving mesial temporal lobe, hippocampal, cerebellar and
cerebral cortex
● Hyperintensities may be seen throughout brain
● Meningeal enhancement may be seen rarely
● Cortical enhancement in absence of restricted diffusion
● PET scan can highlight involvement of mesial temporal lobes
● Normal in 50-60%
43.
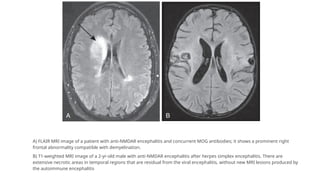
A) FLAIR MRIimage of a patient with anti-NMDAR encephalitis and concurrent MOG antibodies; it shows a prominent right
frontal abnormality compatible with demyelination.
B) T1-weighted MRI image of a 2-yr-old male with anti-NMDAR encephalitis after herpes simplex encephalitis. There are
extensive necrotic areas in temporal regions that are residual from the viral encephalitis, without new MRI lesions produced by
the autoimmune encephalitis
44.
EEG
● EEG isabnormal in virtually all patients
● Focal or diffuse slowing in delta and theta ranges as well as epileptiform discharges
● 30% anti-NMDAR encephalitis exhibit extreme delta brushs characterized by beta–delta complexes
● Continuous combination of delta activity with superimposed fast activity usually in beta region
46.
Antibody testing
● Testingboth serum and CSF is preferred
● Anti-NMDAR CSF testing is more sensitive (100% vs 85%)
● Levels of antibodies in CSF appear to correlate better with outcome
● In protracted disease, delayed diagnosis and after IVIG/PE antibodies may be present only in CSF
47.
● MRI abdomenand pelvis OR Abdominal & Scrotal ultrasound can be done to screen for
tumors
● In cases of negative initial screening, follow-up MRI abdomen and pelvis should be
repeated every six months for at least four years
48.
TREATMENT
● Primary immunomodulationinclude steroids, IVIG or plasma exchange
● Tumor removal if applicable
● Followed by maintenance therapy- oral steroid taper, monthly pulse steroids or pulse IVIG therapy
● Azathioprine and MMF are used in maintenance therapy as steroid sparing agents
● Duration of maintenance therapy ranges from 6 to 12 months
● 2nd
line therapy in case of non response to 1st
line agents: Rituximab, Cyclophosphamide
● 3rd
line agents – Bortezumab, Tocilizumab
49.
First line
● Corticosteroidsare cornerstone of treatment
● Methylprednisolone 30mg/kg/day(max 1g/day) for 3-5 days f/b oral steroids (prednisolone 1-
2mg/kg/day) and taper over 6-12 months
● IVIG (2g/kg over 5 days) or Plasma Exchange(5-7 exchanges of 50ml/kg every alternate day)
● Only 50 % responds to first line treatment
50.
Second line
● Rituximab
●Dose: 375/m2 weekly for 4 weeks IV OR 750 mg/m2 (maximum 1 g) IV twice separated by 2 weeks
● Resulting in B cell depletion and reduced proinflammatory CD4+ and CD8+ T cells
● B cell counts after 2-4 weeks and 3-6 months
● Cyclophosphamide
● Dose: monthly IV infusions 500-1000 mg/m2 BSA for 6-9 months
● Limitations: risks of infertility and secondary malignancies depending on cumulative dose
51.
Third line
● Bortezumab(protease inhibitor- inhibits proinflammatory signaling cascade)
● Tocilizumab (anti-IL-6)
● Intrathecal steroids and MTX
52.
Prognosis:
Mortality Rate- 5%
Recovery(substantially/fully)- 80%
•May take as long as 2 years after symptoms onset
•Last symptoms to improve social interaction, language, executive function
Relapse- 15%
Milder than initial episodes, response well to immunotherapy
Efficacy of chronic immunosuppressant (azathioprine,MMF) in preventing relapse is unknown
53.
Encephalitis with Abagainst GABA-A Receptor:
●Rare AE, that can affect children.
●In adults may occur with thymoma.
●Present with status epilepticus, refractory seizure.
●MRI (T2/FLAIR) brain shows- multifocal hyperintense abnormality.
●Treatment- Immunotherapy & removal of tumor.
54.
Ophelia Syndrome
●Occur ina/w Hodgkin lymphoma.
●Predominantly affect young adults, teenager & children.
●Some develops Ab against mGluR5 receptor involved in learning & memory.
●Management:
1. Removal of tumor
2. Immunotherapy
55.
Hashimoto Encephalopathy:
●Steroid responsiveencephalopathy with autoimmuno-thyroiditis.
●Detection of TPO Ab in patients with acute/subacute encephalitis.
●Detection of TPO Ab is a marker of autoimmunity rather than a disease specific
marker.
●So testing of more relevant Ab like NMDAR Ab should be done.
56.
● Stroke-like symptoms
●Tremor, myoclonus
● Transient aphasia
● Sleep and behaviour abnormalities
● Hallucinations, seizures and ataxia.
⮚ EEG is usually abnormal
⮚ MRI brain- normal
⮚ CSF shows pleocytosis with elevated protein
● TPO antibodies should be viewed as a marker of autoimmunity rather than a
neurologic disease-specific or pathogenic antibody.
57.
⮚ Relevant antibody-associateddisorders, such as GABA(B), LGI1, or
NMDA receptor antibodies
⮚ In some instances, plasma exchange or intravenous
immunoglobulin has been equally effective as steroids.
58.
Bickerstaff Encephalitis:
●Rapid progression(<4wks) of
1. B/L external ophthalmoplegia
2. Ataxia
3. Decreased level of consciousness
●Mostly affect adults, but children of 3 yrs old have been identified.
●Serum GQ1b IgG Ab found in 66% cases.
●May develop hyporeflexia and overlap with MFS.
●MRI T2 signal abnormality (30%)- Brainstem, thalamus & cerebellum.
●Good response to immunotherapy.
59.
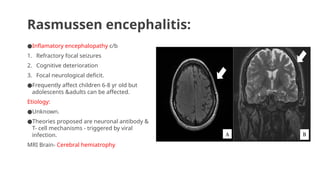
Rasmussen encephalitis:
●Inflamatory encephalopathyc/b
1. Refractory focal seizures
2. Cognitive deterioration
3. Focal neurological deficit.
●Frequently affect children 6-8 yr old but
adolescents &adults can be affected.
Etiology:
●Unknown.
●Theories proposed are neuronal antibody &
T- cell mechanisms - triggered by viral
infection.
MRI Brain- Cerebral hemiatrophy
60.
Management of Rasmussenencephalitis:
1. High dose steroid, IVIG or plasma exchange.
2. Rituximab & intraventricular Gamma interferon effective in isolated cases.
3. Tacrolimus- better outcome of neurological function & slower progression of
cerebral atrophy but no effect on seizure control.
4. Adalimumab( monoclonal antibody against TNF-alpha) shows seizure control
&preservation of cognitive function in 50% cases.
5. Most effective treatment for controlling seizure is functional hemispherectomy
i.e. surgical disconnection of affected hemisphere.
61.
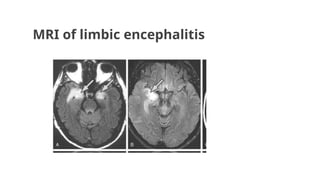
Autoimmune limbic encephalitis:
●Inflamatoryprocess of the limbic system including medial temporal lobe, amygdala, cingulate gyrus.
●Most commonly found in adults.
●Some patients there is underlying tumor i.e. leukemia, ganglioneuroblastoma, neuroblastoma, small cell
carcinoma of ovary.
●Autoantibodies are-
1. VGKCs /LGI1(Leucine rich glioma inactivated 1)
2. Caspr2 antibodies
Clinical features are-
● Severe short term memory loss
● Hyponatremia
● Seizures-myoclonic-like movements (faciobrachial dystonic seizures)
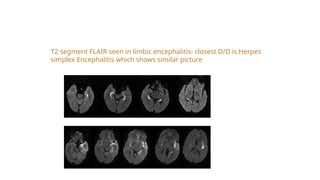
T2 segment FLAIRseen in limbic encephalitis- closest D/D is Herpes
simplex Encephalitis which shows similar picture
64.
◼Other types canoccur with antibodies against intracellular
antigens (eg, Hu, GAD65, Ma2) or against cell surface or
synaptic proteins.
◼Involves T cell immune mediated reactions
◼Responds well to immunotherapy unlike the conventional
form
65.
Opsoclonus-myoclonus and othertype of
brainstem-cerebellar encephalitis:
●In children- 50% have underlying NEUROBLASTOMA.
●In teenagers and young adults underlying TERATOMA usually in ovaries.
●Initially present with- irritability, ataxia, falling, myoclonus, tremor and drooling.
●Later- hypotonia, Opsoclonus c/b rapid, chaotic, multidirectional eye movement without saccadic
interval.
●Immunotherapy improves abnormal eye movement but residual behavioral, language, cognitive
problems persists.
●Delay in treatment appears to be a/w poor outcome , therefore in case of neuroblastoma removal of the
tumor should not delay the start of immunotherapy.
66.
CLIPPERS:
●Chronic Lymphocytic Inflammationwith Pontine Perivascular Enhancement Responsive to Steroids
●Pontine predominant encephalomyelitis
●Usually present with episodic diplopia and facial parasthesia.
●MRI Brain – symmetric curvilinear gadolinium enhancement around the pons & extend variably into
medulla, cerebellum, midbrain and spinal cord.
●Responds to high dose of steroids , may worsen during steroid tapering, requiring chronic steroid.
Brainstem & spinal
cord dysfunction
67.
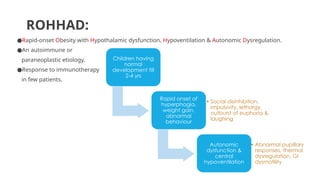
ROHHAD:
●Rapid-onset Obesity withHypothalamic dysfunction, Hypoventilation & Autonomic Dysregulation.
●An autoimmune or
paraneoplastic etiology.
●Response to immunotherapy
in few patients.
Children having
normal
development till
2-4 yrs
Rapid onset of
hyperphagia,
weight gain,
abnormal
behaviour
• Social disinhibition,
impulsivity, lethargy,
outburst of euphoria &
laughing
Autonomic
dysfunction &
central
hypoventilation
• Abnormal pupillary
responses, thermal
dysregulation, GI
dysmotility
68.
NMOSD [NEUROMYELITIS OPTICASPECTRUM DISORDER ]
●Typical involvement of optic nerve & spinal cord.
●Encephalopathy in the form of diencephalic or area postrema syndromes.
●Can have AQP4 or MOG Ab. Some are seronegative.
●Involvement of brain areas rich in AQP4 (periaqueductal gray matter,
hypothalamus, optic nerve and central involvement of the spinal cord).
●High risk of relapses and long term disability.
●Requires chronic immunotherapy.
●Patients with MOG Ab have better outcome than those with AQP4 Ab or
seronegative cases.
























































![NMOSD [NEUROMYELITIS OPTICA SPECTRUM DISORDER ]
●Typical involvement of optic nerve & spinal cord.
●Encephalopathy in the form of diencephalic or area postrema syndromes.
●Can have AQP4 or MOG Ab. Some are seronegative.
●Involvement of brain areas rich in AQP4 (periaqueductal gray matter,
hypothalamus, optic nerve and central involvement of the spinal cord).
●High risk of relapses and long term disability.
●Requires chronic immunotherapy.
●Patients with MOG Ab have better outcome than those with AQP4 Ab or
seronegative cases.](https://image.slidesharecdn.com/autoimmuneencephalitissymposium-1-250913015523-7e4074bf/85/Autoimmune-encephalitis-symposium-1-pptx-68-320.jpg)




































![Circle of willis (finql)[1].pptx anatomy](https://cdn.slidesharecdn.com/ss_thumbnails/circleofwillisfinql1-250913015401-7ca582c5-thumbnail.jpg?width=640&height=640&fit=bounds)






![ONFH[AVN HIP] -TRIPLE REGIME -A NOVAL SURGICAL CONCEPT .pptx](https://cdn.slidesharecdn.com/ss_thumbnails/onfhavnhip2026koaconcalicutdrgokuldevdrmashraf-260210064517-213ec005-thumbnail.jpg?width=640&height=640&fit=bounds)
















