Maj Dr BishwoRaj Kunwar
(Assistant Professor)
Nepalese Army Institute of Health Sciences
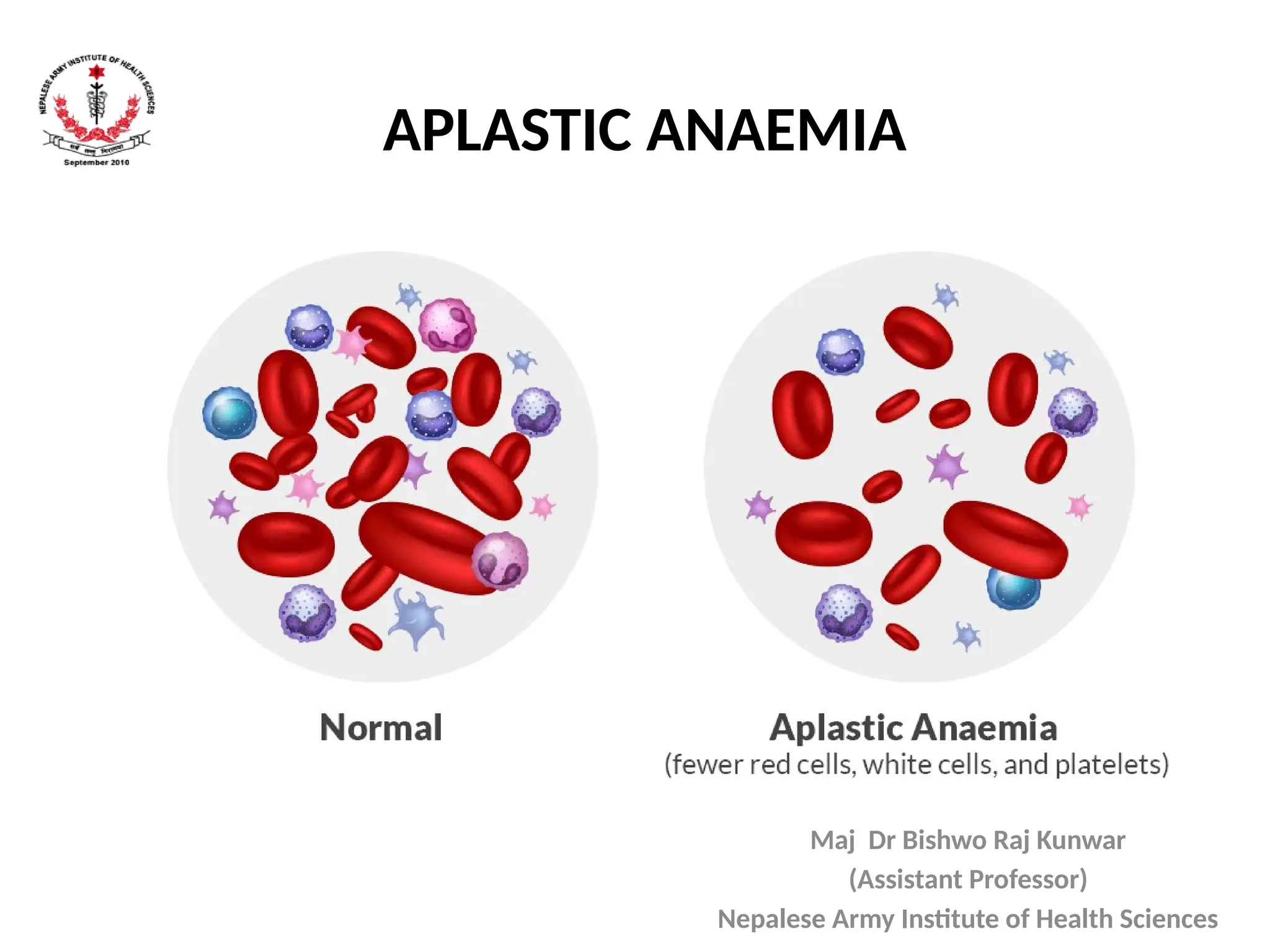
APLASTIC ANAEMIA
2.
Objective
• By theend of this class, students should be
able to:
– Enlist causes of aplastic anemia
– Enumerate clinical findings
– Enlist investigations and treatment measures of
aplastic anemia
3.
VIGNETTE
• A 9-year-oldboy is brought to the pediatric outpatient
clinic with complaints of persistent fatigue, repeated
nosebleeds, and occasional gum bleeding for the past
3 weeks.
• His mother also noticed that he has been developing
bruises on his arms and legs without any history of
trauma. He has no history of fever, weight loss, join
swelling, or recent infections.
4.
VIGNETTE
• On examination:
•Pale conjunctiva
• Multiple ecchymotic patches over the limbs
• No lymphadenopathy
• No hepatosplenomegaly
5.
VIGNETTE
Investigations:
• CBC:
– Hemoglobin:6.2 g/dL (low)
– WBC: 3,200/mm³ (low)
– Platelets: 25,000/mm³ (low)
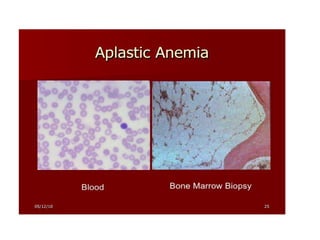
• Peripheral smear: Normocytic, normochromic
anemia, reduced leukocytes and platelets, no
abnormal cells
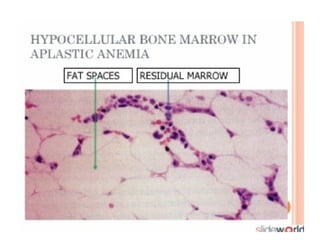
• Bone marrow aspiration: Hypocellular marrow with
marked reduction in all cell lines
6.
VIGNETTE
QUESTIONS:
• What isthe most likely diagnosis?
• Which investigation confirmed the diagnosis?
• List two common causes of aplastic anemia in children.
• Mention two clinical features seen in aplastic anemia.
• Name two important differential diagnoses for this
case.
• Outline one principle of management for this patient.
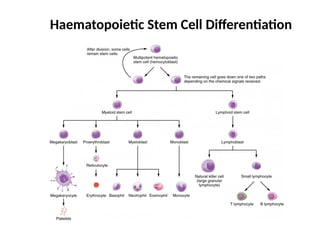
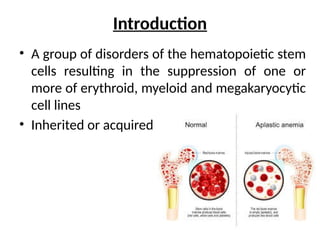
Introduction
• A groupof disorders of the hematopoietic stem
cells resulting in the suppression of one or
more of erythroid, myeloid and megakaryocytic
cell lines
• Inherited or acquired
10.
Etiopathogenesis
• Deficient hematopoieticstem cells due to
– Acquired injury from viruses, toxins or chemicals, or
– Abnormal marrow microenvironment, or
– Immunologic suppression of hematopoiesis (mediated
by antibodies or cytotoxic T cells), or
– Mutations in genes controlling hematopoiesis
(resulting in inherited bone marrow failure syndromes)
Bone marrow failure
11.
History
• Family historyof neonatal thrombocytopenia
(Inherited bone marrow failure syndrome)
• History of exposure to toxins, drugs like
Chloramphenicol, environmental hazards
• History of viral infections (Hepatitis B & C, EBV,
Parvovirus B19)
12.
Epidemiology (Global &Asia)
• Incidence: 2–6 per million/year
• Higher rates in Asia (3–4x more common)
• Common in teenagers and young adults
• No significant gender difference
13.
Prevalence in Nepal
•No national registry
• Case series from Kanti Children's Hospital:
rising detection
• Increased awareness, better diagnostics
• Resource constraints in rural areas
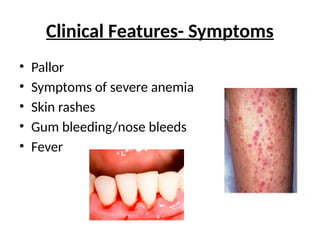
Clinical Features- Signs
•Pallor
• Fever (neutropenia)
• Signs of Congestive heart failure (severe anemia)
• Ecchymosis, petechiae (thrombocytopenia)
• Characteristic congenital physical anomalies
(Inherited bone marrow failure syndrome)
• Signs of infection (pneumonia/sepsis)
• No Lymphadenopathy/ Hepatosplenomegaly
16.
Red Flags inChildren
• Sudden fatigue, frequent infections
• Easy bruising or bleeding
• Recurrent fever
• Non-resolving anemia despite iron therapy
17.
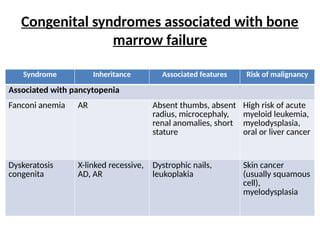
Congenital syndromes associatedwith bone
marrow failure
Syndrome Inheritance Associated features Risk of malignancy
Associated with pancytopenia
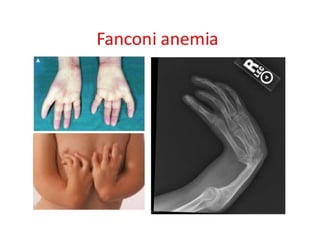
Fanconi anemia AR Absent thumbs, absent
radius, microcephaly,
renal anomalies, short
stature
High risk of acute
myeloid leukemia,
myelodysplasia,
oral or liver cancer
Dyskeratosis
congenita
X-linked recessive,
AD, AR
Dystrophic nails,
leukoplakia
Skin cancer
(usually squamous
cell),
myelodysplasia
19.
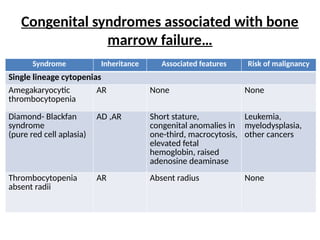
Congenital syndromes associatedwith bone
marrow failure…
Syndrome Inheritance Associated features Risk of malignancy
Single lineage cytopenias
Amegakaryocytic
thrombocytopenia
AR None None
Diamond- Blackfan
syndrome
(pure red cell aplasia)
AD ,AR Short stature,
congenital anomalies in
one-third, macrocytosis,
elevated fetal
hemoglobin, raised
adenosine deaminase
Leukemia,
myelodysplasia,
other cancers
Thrombocytopenia
absent radii
AR Absent radius None
Treatment…
• Criteria forreferral for HSCT:
– Young age
– Severe aplastic anemia
– Availability of matched sibling
• Antithymocyte globulin (ATG):
– Patients with severe acquired aplastic anemia who cannot
undergo HSCT may benefit from ATG along with oral
cyclosporine
• Granulocyte- colony stimulating factor (G-CSF):
– Patients with neutropenia and infection should receive a trial
of G-CSF
– Should be discontinued after seven days (risk of malignant
transformation)
29.
Prognosis
• Determined byseverity and extent of
cytopenias
• Patients with severe aplasia at risk of:
– high output cardiac failure due to anemia;
– bacterial & fungal infections due to neutropenia;
– and severe bleeding due to thrombocytopenia
• With current transplantation regimen, long-
term survival in patients with severe aplastic
anemia 60-70%, with survival rates exceeding
80% in favorable subgroups.
30.
Recent Advances
• Eltrombopag(TPO receptor agonist):
enhances hematopoiesis
• Gene therapy trials (in Fanconi anemia)
• Improved transplant conditioning protocols
• Early use of triple therapy: ATG + CyA +
Eltrombopag
31.
Follow-up Care
• MonthlyCBC monitoring
• Iron overload monitoring (Ferritin)
• Screen for late effects: cancer, marrow failure
relapse
• Psychosocial support and counseling
32.
Hypoplastic Anemia (Differentiation)
•Milder form of marrow suppression
• Often viral or transient (e.g., Parvovirus B19)
• May resolve spontaneously
• Does not usually require transplant
33.
Questions & Answers
•What is the most likely diagnosis?
→ Aplastic anemia.
• Which investigation confirmed the diagnosis?
→ Bone marrow aspiration showing hypocellularity with
decreased all hematopoietic cell lines.
• List two common causes of aplastic anemia in children.
→ Idiopathic (most common)
→ Viral infections (e.g., hepatitis viruses, Epstein–Barr
virus)
→ (Other acceptable: drug-induced, Fanconi anemia).
34.
Questions & Answers
•Mentiontwo clinical features seen in aplastic anemia.
→ Pallor (due to anemia)
→ Bleeding manifestations such as petechiae, purpura, epistaxis
(due to thrombocytopenia)
→ (Also acceptable: recurrent infections due to neutropenia).
•Name two important differential diagnoses for this case.
→ Acute leukemia
→ Megaloblastic anemia
•Outline one principle of management for this patient.
→ Supportive care (packed RBC transfusion, platelet transfusion),
infection control, avoidance of offending drugs, and definitive
therapy with immunosuppressive therapy or hematopoietic stem
cell transplantation in suitable cases.
35.
Objective
• By theend of this class, students should be
able to:
– Enlist causes of aplastic anemia
– Enumerate clinical findings
– Enlist investigations and treatment measures of
aplastic anemia
36.
References
• Nelson Textbookof Pediatrics, 21st Ed
• Kanti Children’s Hospital Clinical Reports
• Nepal Journal of Hematology and Oncology
• WHO Global Health Observatory
• NIH Clinical Guidelines
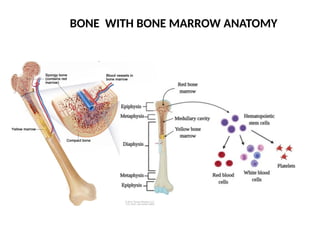
#7 bone : compact bone, spongy bone (proximal and distal end- containing red marrow), bone marrow at the core – bone marrow(central medullary cavity) has red and yellow marrow, red marrow has hematopoietic stem cells , yellow marrow forms: cartilage, fat .(note red marrow is present in both central medullary cavity or spongy bone in children, unlike that of adult)
#10 Autoreactive T cells attacking bone marrow stem cells, leading to bone marrow failure and pancytopenia. Bind to T lymphocyte surface antigens CD2,3,4,8, triggers complement mediated lysis and apoptosis of T cells, reduction of immune-mediated destruction of BM progenitor cells.improves blood counts in 3-6 months.
#22 Macrocytosis is the enlargement of red blood cells
Agranulocytosis is deficiency of granulocytes in the blood, causing increased vulnerability to infection.
#25 Chromosomal breakage study: PBS show a characteristic hypersensitivity towards chromosomal breakage when incubated with DNA cross-linking agents such as Mitomycin C.
Diagnosis of PNH shows that the suspected patient’s red cells have a high sensitivity to complement mediated hemolysis. Partial hemolysis occurs with hereditary erythroblastic multinuclearity disease. Positive test result shows lysis of Red cells in acidified serum samples with patients cell (not with normal cells).
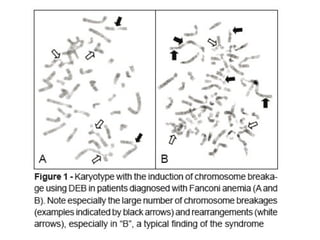
#26 Diepoxybutane: an alkylatic agent it forms DNA-DNA interstrand cross links,which block replication forks.peripheral blood lymphocytes are cultured with and without DEB, so in normal cells, only minimal breakage seen as FANC gene repairs DNA interstrand crosslinks unlike in FA.
#27 Thymocyte : means immature T cells that’s developing inside the thymus. ATG made by immunizing animals with human thymocytes.
G-CSF : a glycoprotein growth factor – a naturally occurring cytokine in your body, it stimulates BM to produce and relase more granulocytes, esp. neutrophils
#28 Fanconi anemia: HSCT is the only cure. Oral androgens have been used as palliative therapy.
ATG : a immunosuppressive agent made from antibodies against human T lymphocytes.derived from horse or rabbit antiboides.
#30 Eltrombopag (TPO receptor agonist : it’s a drug that mimics thrombopoietin ,the natural hormone that stimulates platelet production. Binds to TPO receptors on BM megakaryocyte precursors.


















































































![CTEV [ clubfoot] DR ARUN LAL ,DR MOHAMED ASHRAF travancore medical college k...](https://cdn.slidesharecdn.com/ss_thumbnails/ctevclubfootdrarunlaldrmohamedashraftravancoremedicalcollegekollamkeralaindia-260208063247-18fc466c-thumbnail.jpg?width=640&height=640&fit=bounds)

![ONFH[AVN HIP] -TRIPLE REGIME -A NOVAL SURGICAL CONCEPT .pptx](https://cdn.slidesharecdn.com/ss_thumbnails/onfhavnhip2026koaconcalicutdrgokuldevdrmashraf-260210064517-213ec005-thumbnail.jpg?width=640&height=640&fit=bounds)

