Download to read offline




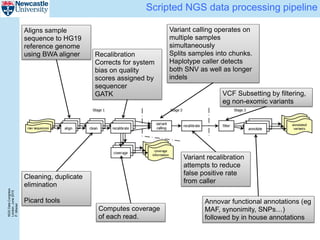
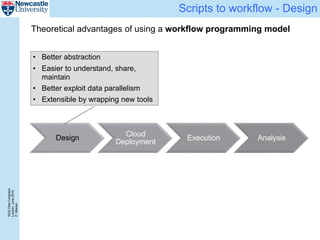
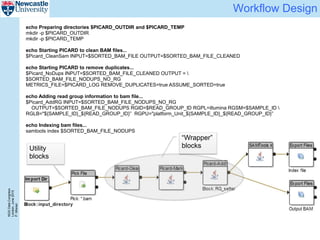
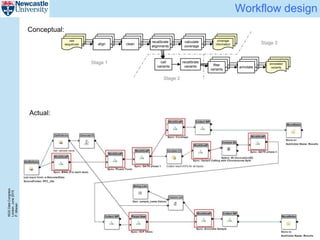
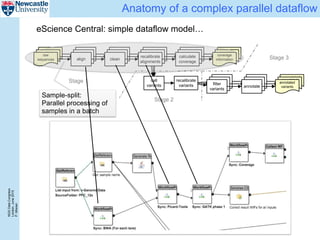
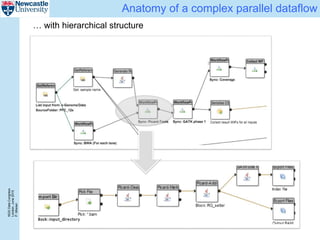
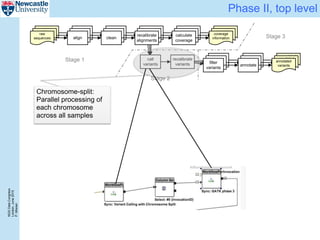
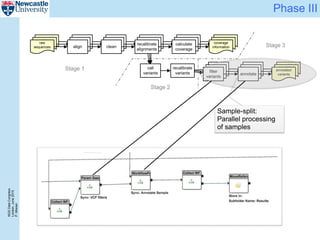
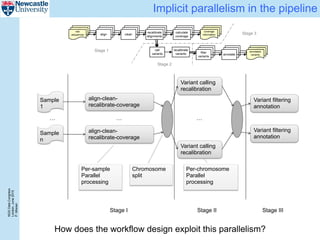
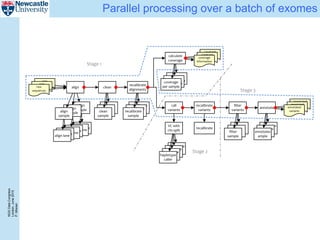
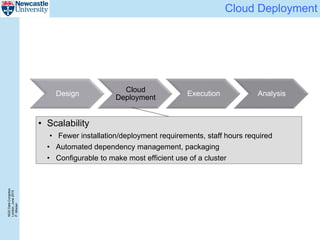
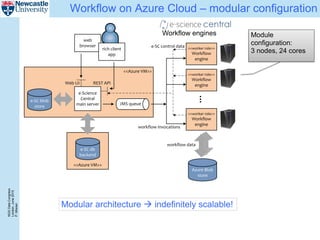
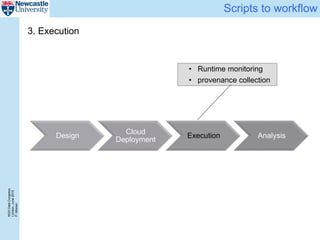
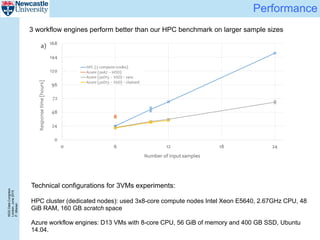
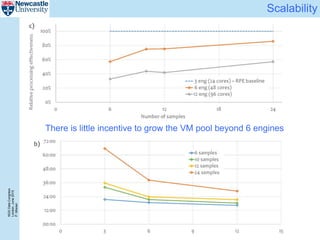
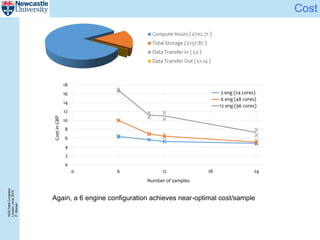
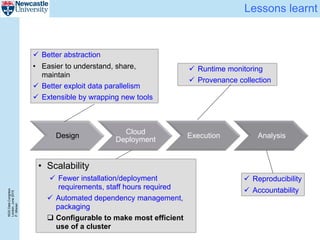
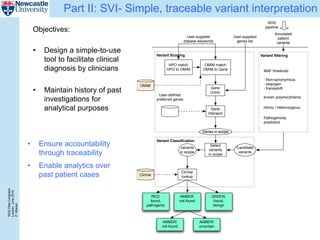
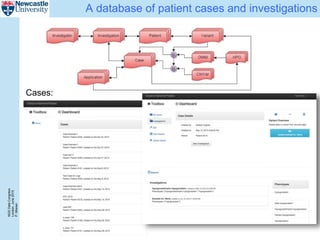
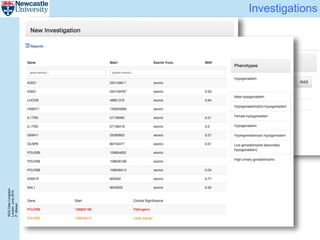
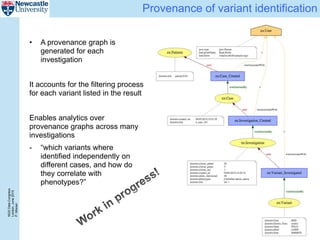
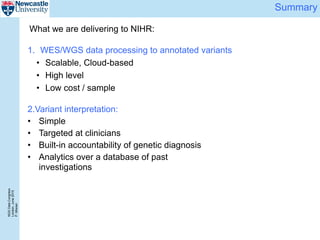

The document describes a scalable WES/WGS processing pipeline and variant interpretation tool. The pipeline uses a workflow programming model to process data in parallel across cloud resources for improved scalability and cost efficiency. The variant interpretation tool provides a simple interface for clinicians while maintaining a history of investigations and provenance for accountability and analytics across patient cases.























































































