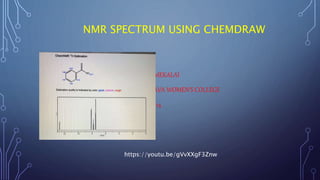
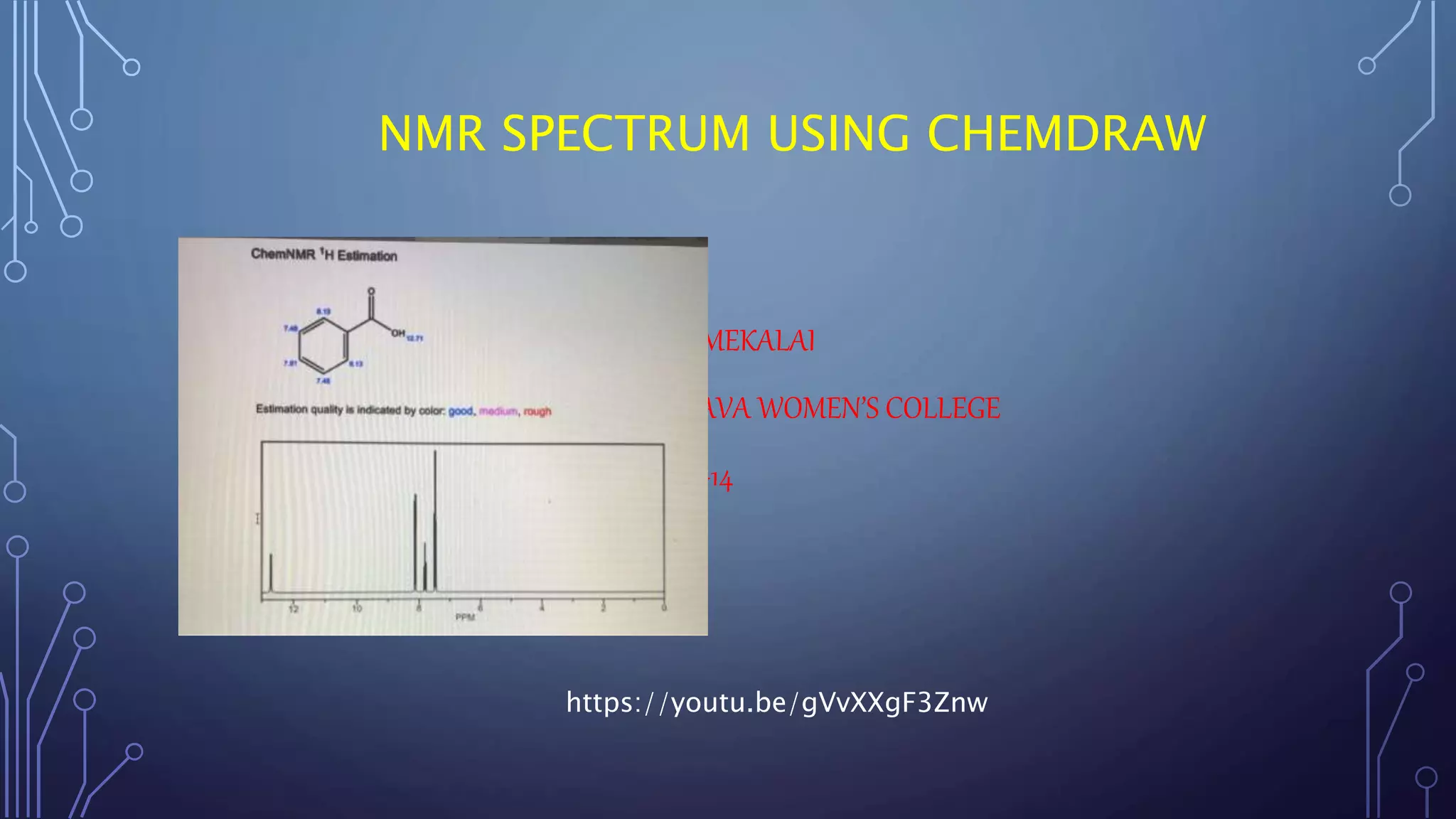
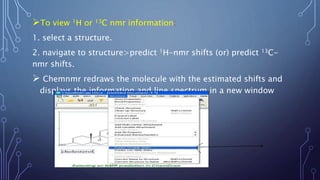
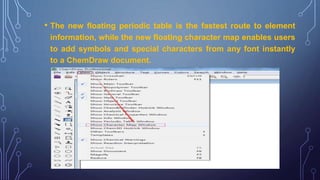
This document discusses using Chemdraw's CS ChemNMR Pro facility to estimate 1H and 13C NMR chemical shifts. It can rapidly compute chemical shifts for large molecules by using fixed parameters for atom types and subgroups. However, it is most reliable for ordinary organic molecules and may be inaccurate for unusual systems. The document provides an example prediction and discusses limitations, such as not estimating shifts for hydrogen atoms bonded to heteroatoms. It also describes assigning structures to spectra and viewing/removing assignments in Chemdraw.























![Noesy [autosaved]](https://cdn.slidesharecdn.com/ss_thumbnails/noesyautosaved-200728183752-thumbnail.jpg?width=640&height=640&fit=bounds)



































































