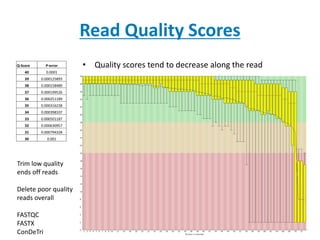
The document discusses next-generation sequencing (NGS) data formats and analyses, detailing the evolution from first to third generation sequencing techniques. It explains the FASTA and FASTQ formats used for representing nucleotide sequences and their quality scores, including the components of these formats and important identifiers like accession numbers. Additionally, it covers quality scores, their implications, and the impact of sequencing technologies on data accuracy.