
Recommended
More Related Content
What's hot
What's hot (20)
Similar to JBEI highlights November 2019
Similar to JBEI highlights November 2019 (20)
More from LeahFreemanSloan
Recently uploaded
Recently uploaded (20)
JBEI highlights November 2019
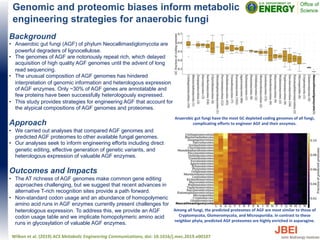
- 1. Genomic and proteomic biases inform metabolic engineering strategies for anaerobic fungi Background • Anaerobic gut fungi (AGF) of phylum Neocallimastiglomycota are powerful degraders of lignocellulose. • The genomes of AGF are notoriously repeat rich, which delayed acquisition of high quality AGF genomes until the advent of long read sequencing. • The unusual composition of AGF genomes has hindered interpretation of genomic information and heterologous expression of AGF enzymes. Only ~30% of AGF genes are annotatable and few proteins have been successfully heterologously expressed. • This study provides strategies for engineering AGF that account for the atypical compositions of AGF genomes and proteomes. Approach • We carried out analyses that compared AGF genomes and predicted AGF proteomes to other available fungal genomes. • Our analyses seek to inform engineering efforts including direct genetic editing, effective generation of genetic variants, and heterologous expression of valuable AGF enzymes. Outcomes and Impacts • The AT richness of AGF genomes make common gene editing approaches challenging, but we suggest that recent advances in alternative T-rich recognition sites provide a path forward. • Non-standard codon usage and an abundance of homopolymeric amino acid runs in AGF enzymes currently present challenges for heterologous expression. To address this, we provide an AGF codon usage table and we implicate homopolymeric amino acid runs in glycosylation of valuable AGF enzymes. Wilken et al. (2019) ACS Metabolic Engineering Communications, doi: 10.1016/j.mec.2019.e00107 Among all fungi, the predicted proteomes of AGF are most similar to those of Cryptomycota, Glomeromycota, and Microsporidia. In contrast to these neighbor phyla, predicted AGF proteomes are highly enriched in asparagine. Anaerobic gut fungi have the most GC depleted coding genomes of all fungi, complicating efforts to engineer AGF and their enzymes.
- 2. Function-driven single-cell genomics uncovers cellulose-degrading bacteria from the rare biosphere Background • Assigning a functional role to a microorganism has historically relied on cultivation of isolates or detection of environmental genome-based biomarkers using a posteriori knowledge of function. However, the emerging field of function-driven single-cell genomics aims to expand this paradigm by identifying and capturing individual microbes based on their in situ functions or traits. Approach • We developed and benchmarked a function-driven single-cell screen for cellulase activity in uncultivated microbes, which we applied to a microbial community inhabiting the Great Boiling Spring (GBS) Geothermal Field, northwest Nevada. Outcomes and Impacts • Single bacterial cells that bound to the fluorescent cellulose probe were sorted and characterized by 16S rRNA and single cell sequencing. • Putative cellulases were identified by bioinformatics and screened using an in vitro assay and NIMS detection. • Along with well-characterized phyla, divergent cellulases encoded in the genome of a representative of a recently described candidate phylum from the rare biosphere, Goldbacteria, were identified and characterized. • We expect that this function-driven single-cell approach can be extended to a broad range of substrates, linking microbial taxonomy directly to in situ function.. Doud et al. (2019) ISME J, doi:10.1038/s41396-019-0557-y
- 3. Evaluating protic ionic liquid for woody biomass one-pot pretreatment + saccharification, followed by Rhodosporidium toruloides cultivation Background • One-pot ionic liquid-based processes are a promising bioproduction strategy to reduce water consumption (removing the washing of biomass), reducing costs and minimizing the presence of inhibitory compounds. Approach • We evaluated the one-pot ionic liquid pretreatment + saccharification configuration as a scheme for the deconstruction and conversion of two different woody feedstocks, eucalyptus, and pine. • A comparative study of protic ionic liquids 2- hydroxylethylammonium acetate-based ionic liquids, bis-2- hydroxyethyl ammonium acetate ([2-HEA][OAc]), and cholinium lysinate ([Ch][Lys]) for pretreatment of eucalyptus and pine was conducted. Outcomes and Impacts • Protic ionic liquids were more effective in eucalyptus than in pine pretreatment and less toxic than cholinium lysinate at concentrations ≤10 % w/w. • 2-hydroxylethylammonium acetate yielded the highest digestibility, of up to 75% in eucalyptus. • This work is a starting point for further studies aimed at increasing cellulose digestibility in a one-pot configuration in the presence of protic ILs. Rigual et al. (2019) ACS Sustainable Chem. Eng., doi:10.1021/acssuschemeng.9b04451 Effect of pH adjustment on glucose and xylose consumption over the course of growth and production using R. toruloides in the presence of IL [2-HEA][OAc].
- 4. Omics-driven identification and elimination of valerolactam catabolism in Pseudomonas putida KT2440 for increased product titer Background • Pseudomonas putida is a promising host for metabolic engineering due to its diverse catabolic range allowing for the valorization of lignin derivatives. • P. putida is also able to degrade or catabolize multiple lactams, important precursors to polymers such as nylon. This catabolism dramatically impacts titers in engineered strains. Approach • We utilized a combination of Random-Barcode Transposon Sequencing (RB-TnSeq), as well as shotgun proteomics to identify enzymes that hydrolyze lactams in P. putida. We then used this information to engineer more productive strains of lactam producing strains of P. putida. Outcomes and Impacts • RB-TnSeq validated that valerolactam is metabolized via the L- lysine catabolic pathway. • Shotgun proteomics identified that OplBA is likely responsible for lactam hydrolysis. • Knocking out oplBA in P. putida prevented the bacterium from growing on valerolactam, as well as preventing caprolactam hydrolysis in vivo. • Knocking out oplBA and other loci in P. putida increased valerolactam titers from 0 mg/L to ~90 mg/L after 48-hour fermentations with added L-lysine. Thompson et al. (2019) Metab Eng Commun. doi: 10.1016/j.mec.2019.e00098 Using a combination of RB-TnSeq and shotgun proteomics a lactam hydrolase was identified in P. putida (A). By eliminating the hydrolase and other pathways that compete for precursors valerolactam titer was significantly increased (B).
- 5. Genome sequence of the model rice variety KitaakeX Background • Rice (Oryza sativa) provides food for more than half of the world’s population and also serves as a model for studies of biofuel crops such as sorghum and switchgrass • The Kitaake cultivar (ssp. japonica), which originated at the northern limit of rice cultivation in Hokkaido, Japan, has emerged as a model for rice research. It has a rapid life cycle (9 weeks seed to seed) and is easy to transform and propagate • KitaakeX, a Kitaake variety carrying the Xa21 immune receptor gene Jain et al. (2019) BMC Genomics, doi:10.1186/s12864-019-6262-4 Genome wide analysis of KitaakeX genome and its comparison with other rice varieties Approach • To obtain a high-quality, de novo genome assembly, we sequenced the KitaakeX genome using a strategy that combines short-read and long-read sequencing. Sequencing reads were collected using Illumina, 10x Genomics, PACBIO, and Sanger platforms at the Joint Genome Institute (JGI) and the HudsonAlpha Institute Outcomes • The assembled sequence contains 377.6 Mb, consisting of 33 scaffolds with a contig N50 of 1.4 Mb, covering a total of 99.67% of assembled bases in chromosomes • We predicted 35,594 protein-coding genes in the KitaakeX genome representing 31.5% genic space of the assembled genome size. • We found 331,335 variations between KitaakeX and Nipponbare and nearly 10 times as many (2,785,991) variations between KitaakeX and Zhenshan97 Significance • The high quality, de novo assembly of the KitaakeX genome will serve as a useful reference genome for rice and will accelerate functional genomics studies of rice and other monocotyledonous species such as sorghum. The early flowering rice variety KitaakeX
- 6. Stochastic economic and environmental footprints of biodiesel production from Jatropha curcas Linnaeus in Nepal Background • Despite some initial failures, the required growing conditions for commercial success of Jatropha have now identified, which lead to a renewed commercial interest and necessitate a new and updated analysis. • This study considers a novel set of essential parameters specifics to geography, climate, soil conditions, and irrigation to determine commercially feasible land for Jatropha farming and documented a system level economic and environmental impacts analyses. Approach • We developed stochastic process models and quantified selling price and carbon footprint. • We determined suite of avenues to address the current global failure of Jatropha-based biodiesel plant. Outcomes and Impacts • A seed yield of >3.9 t/ha and a high oil content Jatropha variety (oil yield of >50 wt%) are required to achieve the selling price of biodiesel close to the current local price of the conventional diesel of $1/L. • Including the impacts from direct and indirect land use changes, the carbon footprint could reach below the conventional fuel equivalent by achieving a very high seed yield (>5 t/ha), using only marginal lands, and encouraging aggressive afforestation. • Results indicate the pathways and sensitivities for developing policies to enable the production of sustainable biodiesel from Jatropha. Baral et al. (2019) Renew. Sustain. Energy Rev., doi: 10.1016/j.rser.2019.109619 Minimum selling price of biodiesel Greenhouse gas emissions including direct land use changes