Downloaded 29 times




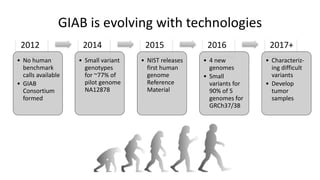


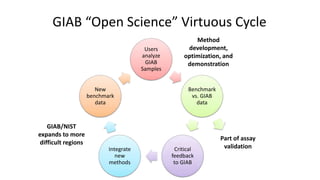

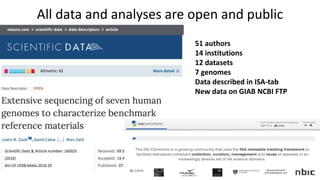
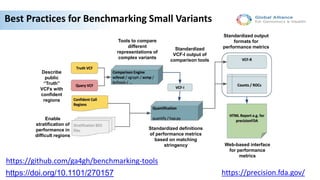
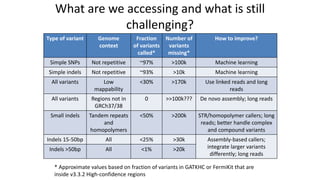
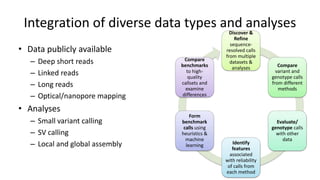

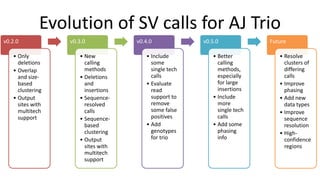
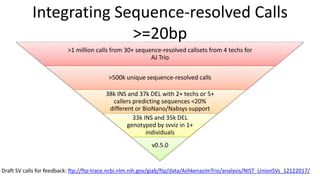
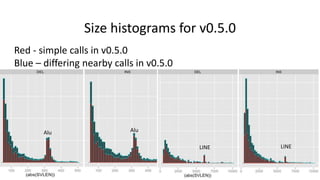






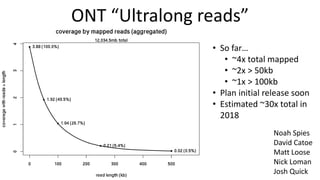

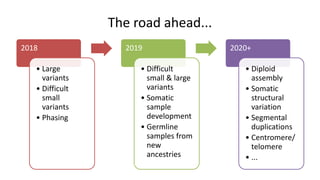




Genome in a Bottle aims to provide well-characterized human genomes as benchmarks to validate genome sequencing and variant calling. The summary characterizes five genomes that have been analyzed to provide benchmark calls for simple and some complex variants, though many challenges remain, particularly for structural variants and difficult genomic regions. Integration of multiple data types and analyses from diverse technologies is key to improving benchmark calls over time in an open and transparent manner.





































































![CTEV [ clubfoot] DR ARUN LAL ,DR MOHAMED ASHRAF travancore medical college k...](https://cdn.slidesharecdn.com/ss_thumbnails/ctevclubfootdrarunlaldrmohamedashraftravancoremedicalcollegekollamkeralaindia-260208063247-18fc466c-thumbnail.jpg?width=640&height=640&fit=bounds)

![ONFH[AVN HIP] -TRIPLE REGIME -A NOVAL SURGICAL CONCEPT .pptx](https://cdn.slidesharecdn.com/ss_thumbnails/onfhavnhip2026koaconcalicutdrgokuldevdrmashraf-260210064517-213ec005-thumbnail.jpg?width=640&height=640&fit=bounds)














