Downloaded 834 times



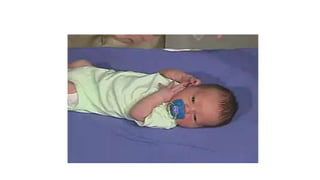
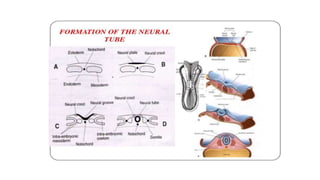

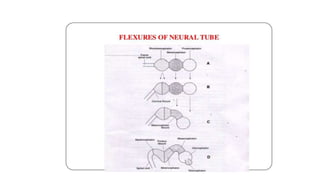




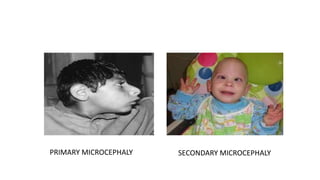



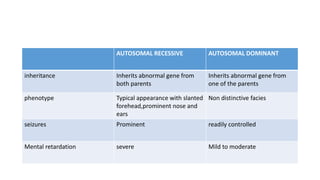




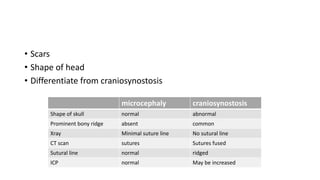






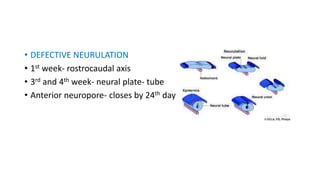

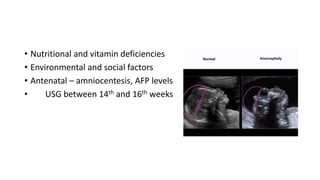
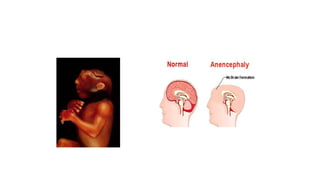
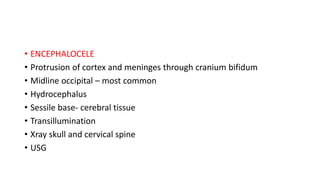

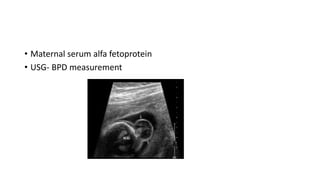
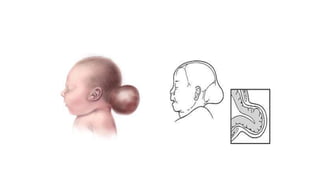
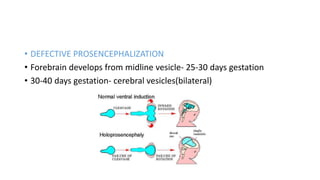
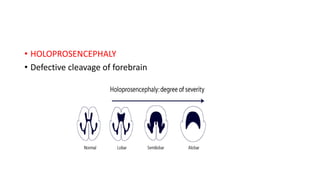




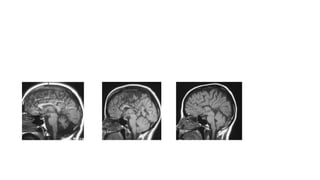
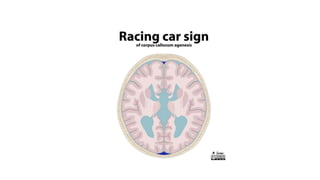
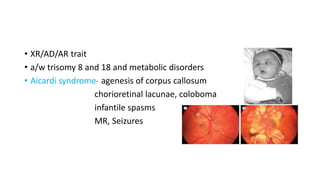
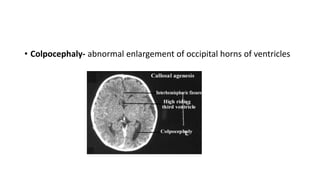
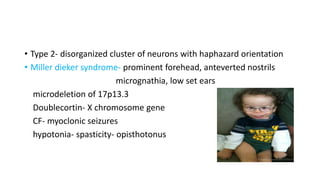



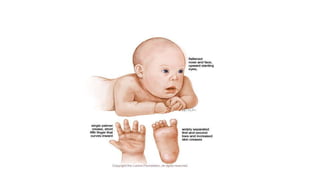














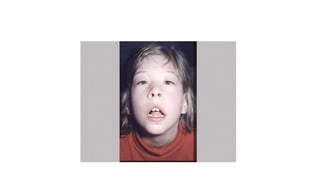









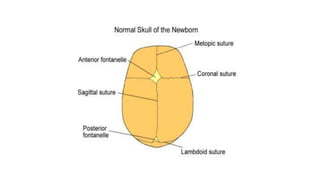

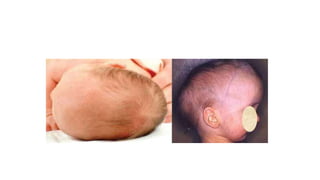



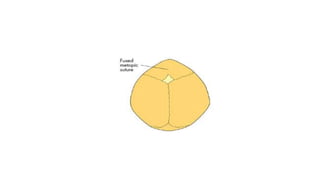

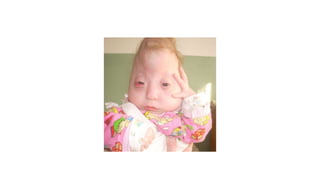

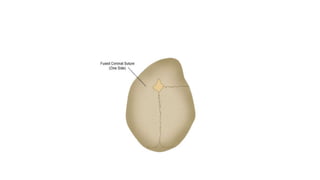

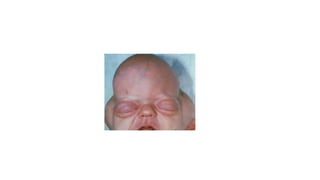



Microcephaly is a head circumference more than 3 standard deviations below the mean. It can be primary/genetic due to defects in cellular migration, neurulation or prosencephalization. Secondary microcephaly has prenatal causes like infections, drugs or postnatal causes like birth injuries or infections. Primary microcephaly is usually autosomal recessive and presents with distinctive facial features and severe intellectual disability. Secondary microcephaly has a varied presentation depending on the cause. Evaluation involves examining for dysmorphism, neurological problems and investigating for possible causes. Treatment focuses on managing symptoms while prevention centers around screening for infections and nutritional supplementation.























































































