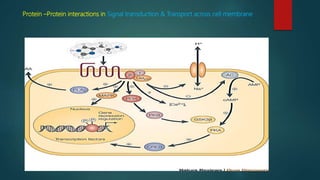
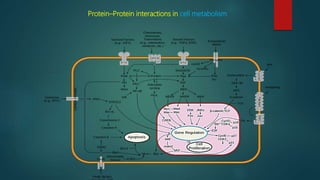
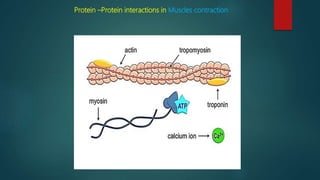
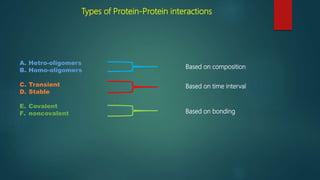
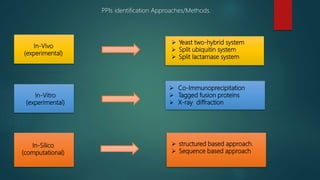
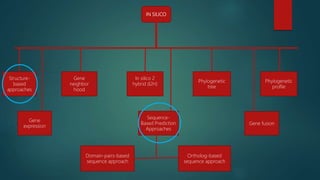
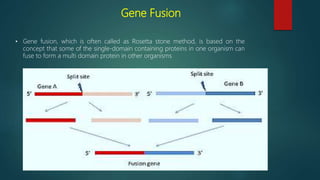
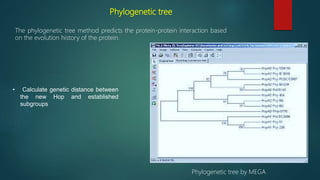
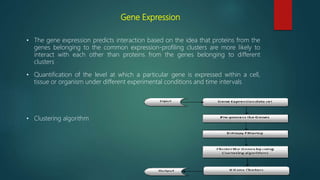
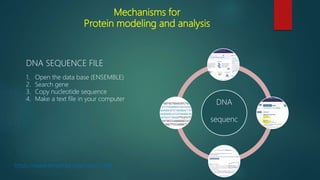
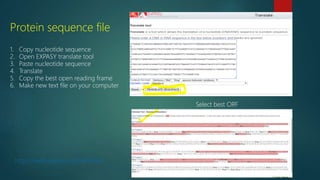
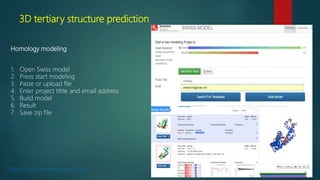
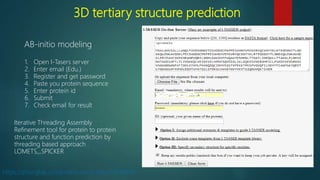
The document discusses protein-protein interactions (PPIs), which occur when two or more protein molecules make physical contact with each other. It describes different types of PPIs such as homo-oligomers and hetero-oligomers, as well as transient and stable interactions. Methods for studying PPIs are also examined, including experimental techniques like yeast two-hybrid systems as well as computational approaches like structure-based modeling and sequence-based prediction. Protein docking is discussed as a way to model and analyze PPIs at the atomic level.