
Download to read offline




























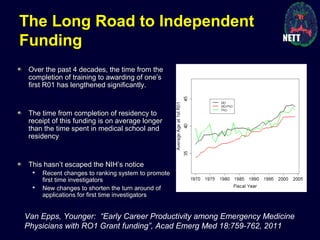



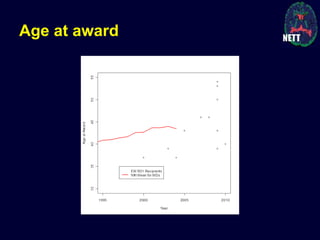
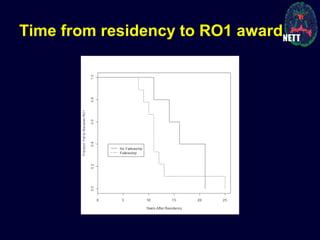
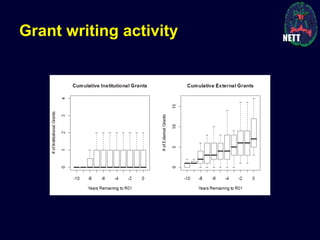









The document discusses the challenges and failures in neuroprotection research, particularly in stroke and traumatic brain injury, highlighting the inadequacies of traditional clinical trial designs and the need for adaptive trial methodologies. It emphasizes the importance of better collaboration in preclinical and clinical research, the redesign of trial processes, and utilizing flexible designs to improve outcomes. Additionally, it touches on the long journey to obtaining research funding, particularly for emergency medicine physicians, and the factors contributing to a successful research career.














































































![Hypothalamus short ppt by Dr. Neha [PT].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/hypothalamusbydr-260124145759-b9f94a93-thumbnail.jpg?width=640&height=640&fit=bounds)
















