Download to read offline







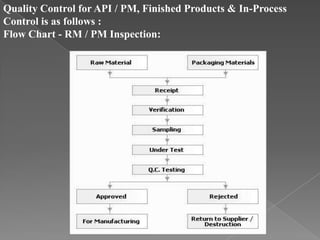
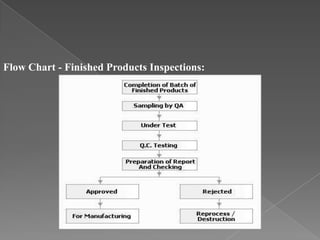

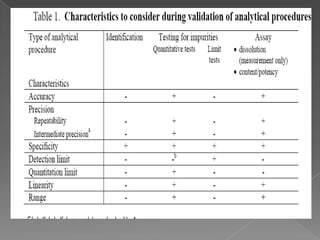
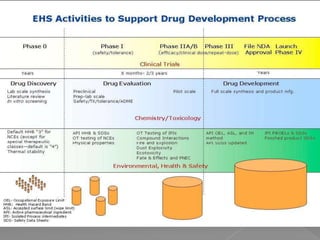



























Quality assurance in medicinal product manufacturing ensures that products meet safety, efficacy, and quality standards through organized systems and practices, including good manufacturing and laboratory practices. The quality system encompasses key areas such as quality control, production, distribution, and inspections, with specific responsibilities assigned to qualified personnel. Documentation and stringent monitoring of processes, equipment, and training are crucial for maintaining compliance and preventing contamination.




















































































